
jak replikować Mitochondię?
replikacja mitochondrialna jest schematyczna w kreskówce w pasku bocznym i pokazana powyżej w mikrografie elektronowym. Mitochondria replikują się podobnie jak komórki bakteryjne. Kiedy stają się zbyt duże, przechodzą rozszczepienie. Wiąże się to z bruzdą wewnętrznej, a następnie zewnętrznej błony, tak jakby ktoś szczypał mitochondrion. Następnie dwie córki mitochondria się rozdzieliły. Oczywiście, mitochondria muszą najpierw replikować swoje DNA. Zostanie to omówione bardziej szczegółowo w następnej sekcji. Elektronowy mikrograf przedstawiający proces bruzd jest pokazany na tych rysunkach. Powyższy rysunek pochodzi z Fawcett, A Textbook of Histology, Chapman and Hall, 12th edition, 1994

czasami nowe mitochondria są syntetyzowane de novo w ośrodkach, które są bogate w białka i polirybosomy potrzebne do ich syntezy. Mikrograf elektronowy na powyższym rysunku pokazuje takie centrum. Wydaje się, że klaster mitochondriów znajduje się w matrycy białek i innych materiałów potrzebnych do ich produkcji. Jak możesz udowodnić, że materiał w tym regionie wytwarzał białka mitochondrialne? Powrót do Menu
niektóre białka mitochondrialne są potrzebne, zanim mitochondria mogą się dzielić.
Wykazano to w badaniu przeprowadzonym przez Sorgo i Yaffe, J Cell Bio. 126: 1361-1373, 1994. Wykazały one Wynik usunięcia białka błony zewnętrznej z mitochondriów o nazwie MDM10. Ta liczba pokazuje wyniki. Mitochondria są w stanie przyjmować składniki i wytwarzać błony i enzymy matrycowe. Jednak rozszczepienie nie jest dozwolone. W rezultacie powstaje gigantyczny mitochondrion. To jest złe w mikrografie poniżej.

mitochondrialne DNA i jego funkcja.
Mitochondria mają własne DNA, rybosomy i mogą wytwarzać wiele własnych białek. DNA jest okrągłe i leży w matrix.in struktury punktowe zwane „nukleoidami”. Każdy nukleoid może zawierać 4-5 kopii mitochondrialnego DNA (mrDNA).
ludzkie DNA mitochondrialne wynosi 16 569 bp; koduje wiele białek mitochondrialnych
- podjednostek 1, 2 i 3 oksydazy cytochromu
- podjednostek 6, 8,9 ATPazy Fo
- podjednostki Apocytochromu b reduktazy coqh2-cytochromu C
- siedem podjednostek reduktazy nadh-Coq
jądro koduje pozostałe białka. Większość lipidów jest importowana(przypomnijmy wykłady na temat dodawania lipidów do błon). Ta kreskówka z twojego tekstu pokazuje udział nuklearny. Podświetlone etykiety to leki, które można wykorzystać do blokowania procesu i testowania źródła białka mitochondrialnego.

Mitochondria mają również własne rybosomy i tRNA:
- 22 tRNAs
- rRNAs
- 16S
- 12S
- 5S
(Magalhaes, PJ; Andreu, AL, Schon EA, dowody na obecność 5 S rRNA w komórkach mitochondriów mol biol ssaków 9: 2375-2382)
rysunek po lewej stronie przedstawia rybosomy mitochondrialne w postaci granulek w mitochondriach.
teksty nadal mówią, że mitochondria nie mają 5s rRNA, jednak ostatnie badania przytoczone powyżej pokazują dowody na 5S w starannie przygotowanych frakcjach mitochondrialnych. Pracownicy ci znaleźli 5S w wysoko oczyszczonych mitochondriach i mitoplastach (mitochondria bez zewnętrznej błony). Wniosek: 5s rRNA jest importowany do mitochondriów, ale jego funkcja jest niepewna.
Wizualizacja mitochondrialnego DNA
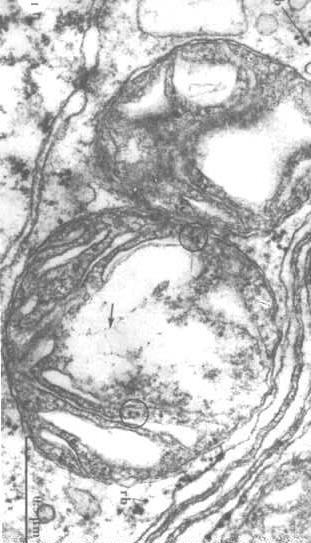
aby zwizualizować  strukturę mitochondrialnego DNA, musimy wyodrębnić białka w matrycy i ujawnić DNA (strzałki na rysunku po prawej stronie).
strukturę mitochondrialnego DNA, musimy wyodrębnić białka w matrycy i ujawnić DNA (strzałki na rysunku po prawej stronie).
można również zobaczyć rybosomy w kręgach.
Alternatywnie, można wyodrębnić DNA i unieść go na powierzchni wody. Następnie można go podnieść za pomocą siatki pokrytej tworzywem sztucznym i zbadać w mikroskopie elektronowym. Mitochondrialne okrągłe DNA przedstawiono na poniższym rysunku. Ten Electron micrograph is taken from Fawcett, A Textbook of Histology, Chapman and Hall, 12th edition, 1994.
dziedziczenie mitochondrialne
u ssaków 99,99% mitochondrialnego DNA (mtDNA) jest dziedziczone po matce. Dzieje się tak, ponieważ plemniki przenoszą swoje mitochondria wokół części ogona i mają tylko około 100 mitochondriów w porównaniu do 100 000 w oocytach. W miarę rozwoju komórek coraz więcej mtDNA od mężczyzn jest rozcieńczanych. Stąd mniej niż jedna część w 104 lub 0,01% mtDNA jest ojcowska. Oznacza to, że mutacje mtDNA mogą być przekazywane z matki na dziecko. Ma to również konsekwencje, jeśli ktoś robi klonowanie ssaków z wykorzystaniem komórek somatycznych. DNA jądrowe pochodzi z komórki dawcy, ale mtDNA pochodzi z komórki gospodarza. Tak sklonowano owcę Dolly.
istnieje szczep drożdży, zwany „Petite”, który ma strukturalnie nieprawidłowe mitochondria, które są niezdolne do fosforylacji oksydacyjnej. Te mitochondria straciły część lub całość swojego DNA. Dziedziczenie mitochondrialne od drożdży jest dwuwarstwowe, a obie komórki macierzyste przyczyniają się do komórek potomnych, gdy komórki haploidalne łączą się. Po mejozie i mitozie następuje losowa dystrybucja mitochondriów do komórek potomnych. Jeśli fuzja jest z drobnymi drożdżami i drożdżami, które nie są, pewien procent komórek potomnych będzie „drobny”.
mutacje w mtDNA ssaków powodują choroby, ponieważ w sekwencji występuje tak krótka sekwencja i bardzo duża zawartość informacji. Następny wykładowca na mitochondriach w tej serii spędzi dużo czasu na genomie mitochondrialnym. Ponieważ każda komórka zawiera setki mitochondriów i tysiące kopii genomu, efekty zmutowanych mitochondriów mogą zostać rozcieńczone. Zgodnie z oczekiwaniami, te tkanki lub narządy, które mogą być najbardziej dotknięte, będą najbardziej zależne od fosforylacji oksydacyjnej (produkcja ATP). U młodych osób może nie zostać odebrany, ponieważ nawet osoba z 15% prawidłowymi mitochondriami może mieć wystarczająco dużo, aby być zdrowym. Jednak starzejący się pacjenci mogą wykazywać cięższy fenotyp choroby.
niektóre przykłady chorób:
- dziedziczna neuropatia nerwu wzrokowego Lebera (zwyrodnienie nerwu wzrokowego, któremu towarzyszy zwiększona ślepota): spowodowana mutacją genu kodującego podjednostkę 4 reduktazy nadh-C0Q.
- „poszarpane włókna mięśniowe” związane z gwałtownymi ruchami są spowodowane mutacją mitochondrialnej lizyny tRNA.
- zespół Kaernsa-Sayre’ a: wady wzroku, nieprawidłowe bicie serca, zwyrodnienie ośrodkowego układu nerwowego. Kilka dużych delecji w mtDNA.
czy można naprawić uszkodzone DNA mitochondrialne?
aktualne badania mówią tak.
- Meeusen, S, Tieu, Q, Wong E, Weiss, e, Schieltz, D, Yates, JR i Nunnari, J. Mgm101p jest nowym składnikiem mitochonrialnego nukleoidu, który wiąże DNA i jest wymagany do naprawy oksydacyjnie uszkodzonego mitochondrialnego DNA. J Cell Biol 145: 291-304 (1999)
- Mgm oznacza „mitochondrial genome maintenance”. Odkryto go w komórkach drożdży podczas poszukiwania mutantów, które powodowały wrażliwą na temperaturę utratę mitochondrialnego DNA.
- Skondensował Mgm101 z Zielonym fluorescencyjnym białkiem i odkrył, że jest zlokalizowane w punktowych strukturach „nukleoidowych”. Lokalizacja pokrywała się z lokalizacją systemów detekcji DNA.
- po wykryciu badania przesiewowego białka Mgm101 zbadano, w jaki sposób jego utrata wpływa na kompetencje oddechowe. Oczywiście białko było potrzebne do funkcjonowania, ale nie wiedzą dokładnie, jaka jest jego rola w tym momencie.
- Sugerowało to, że Mgm101p może mieć zdolność wiązania DNA. Porównano jego wiązanie z kolumnami celulozy DNA (w warunkach wysokiej soli) z innym znanym białkiem wiążącym DNA i potwierdzono stosunkowo wysokie powinowactwo wiązania przez oba białka.
co się dzieje ze starymi, zużytymi mitochondriami?
liczba mitochondriów jest kontrolowana przez autofagię. Jest to proces, w którym lizosomy biorą udział w kontrolowaniu składników komórek. Ta liczba pokazuje proces; pochodzi z Fawcett, Podręcznik histologii, Chapman and Hall, 12th edition, 1994.
proces rozpoczyna się od owinięcia błony retikulum endoplazmatycznego wokół mitochondrium. Następnie pęcherzyki pochodzą z kompleksu Golgiego i łączą się z autofagiczną wakuolą. Pęcherzyki te zawierają hydrolazy przyłączone do receptorów fosforanu mannozy 6 w błonach pęcherzyków. Strona internetowa lysosome omawia ich funkcję i Los. Przypomnijmy, że łączą się z autofagiczną wakuolą. Kwasowe pH umożliwia następnie usunięcie hydrolaz z ich receptorów. Receptory są zawracane z powrotem do kompleksu Golgiego w innych pęcherzykach.
w międzyczasie lizosom tworzy się, gdy spada pH, a komórki zaczynają degradować zawartość. Przypomnijmy, że lizosomy są lampą+, ale nie przenoszą MPR, ponieważ zostały one poddane recyklingowi do kompleksu Golgiego. Jaki płaszcz znajduje się wokół pęcherzyków transportowych prowadzących do autofagicznej wakuoli?
aby uzyskać więcej informacji, skontaktuj się z:
, FAAA
profesor i Katedra
Department of Neurobiology and Developmental Sciences
University of Arkansas for Medical Sciences
Little Rock, AR 72205
w przypadku pytań skontaktuj się z tym adresem e-mail: