
Wie replizieren sich Mitochondien?
Die mitochondriale Replikation ist im Cartoon in der Seitenleiste schematisch dargestellt und oben in einer elektronenmikroskopischen Aufnahme dargestellt. Mitochondrien replizieren sich ähnlich wie Bakterienzellen. Wenn sie zu groß werden, werden sie gespalten. Dies beinhaltet ein Furchen der inneren und dann der äußeren Membran, als würde jemand das Mitochondrium einklemmen. Dann spalteten sich die beiden Tochter-Mitochondrien. Natürlich müssen die Mitochondrien zuerst ihre DNA replizieren. Dies wird im nächsten Abschnitt ausführlicher besprochen. Eine elektronenmikroskopische Aufnahme, die den Furchvorgang darstellt, ist in diesen Figuren gezeigt. Die obige Abbildung wurde von Fawcett genommen, Ein Lehrbuch der Histologie, Chapman und Hall, 12. Auflage, 1994

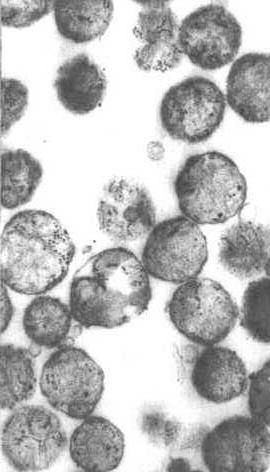
Manchmal werden neue Mitochondrien de novo in Zentren synthetisiert, die reich an Proteinen und Polyribosomen sind, die für ihre Synthese benötigt werden. Die elektronenmikroskopische Aufnahme in der obigen Abbildung zeigt ein solches Zentrum. Es scheint, dass der Cluster von Mitochondrien in einer Matrix von Proteinen und anderen Materialien sitzt, die für ihre Produktion benötigt werden. Wie könnten Sie beweisen, dass Material in dieser Region mitochondriale Proteine herstellte?
Bestimmte mitochondriale Proteine werden benötigt, bevor sich die Mitochondrien teilen können.
Dies wurde in einer Studie von Sorgo und Yaffe, J Cell Bio, gezeigt. 126: 1361-1373, 1994. Sie zeigten das Ergebnis der Entfernung eines äußeren Membranproteins aus Mitochondrien namens MDM10. Diese Abbildung zeigt die Ergebnisse. Die Mitochondrien sind in der Lage, Komponenten aufzunehmen und Membranen und Matrixenzyme zu produzieren. Spaltung ist jedoch nicht erlaubt. Das Ergebnis ist also ein riesiges Mitochondrium. Dies ist illstrated in der mikroskopischen Aufnahme unten.

Mitochondriale DNA und ihre Funktion.
Mitochondrien haben einige ihrer eigenen DNA, Ribosomen, und können viele ihrer eigenen Proteine herstellen. Die DNA ist kreisförmig und liegt in der matrix.in punktförmige Strukturen, die „Nukleoide“ genannt werden. Jedes Nukleoid kann 4-5 Kopien der mitochondrialen DNA (mrDNA) enthalten.
Menschliche mitochondriale DNA ist 16,569 bp; kodiert eine Anzahl von mitochondrialen Proteinen
- Untereinheiten 1, 2 und 3 der Cytochromoxidase
- Untereinheiten 6, 8,9 der Fo-ATPase
- Apocytochrom b-Untereinheit der CoQH2-Cytochrom-C-Reduktase
- Sieben NADH-CoQ-Reduktase-Untereinheiten
Der Zellkern kodiert für die restlichen Proteine. Das meiste Lipid wird importiert (erinnern Sie sich an die Vorlesungen über die Lipidaddition zu Membranen). Dieser Cartoon aus Ihrem Text zeigt die nukleare Beteiligung. Die hervorgehobenen Etiketten sind Medikamente, die verwendet werden können, um den Prozess zu blockieren und die Quelle des mitochondrialen Proteins zu testen.

Mitochondrien haben auch ihre eigenen Ribosomen und tRNA:
- 22 tRNAs
- rRNAs
- 16S
- 12S
- 5S
( Magalhaes, PJ; Andreu, AL, Schon EA, Beweise für das Vorhandensein von 5 S rRNA in Säugetier-Mitochondrien Mol Biol Zelle 9: 2375-2382)
Die Abbildung links zeigt mitochondriale Ribosomen als Granulate in den Mitochondrien.
Die Texte sagen immer noch, dass Mitochondrien keine 5S rRNA haben, aber die kürzlich zitierte Studie zeigt Beweise für 5S in sorgfältig vorbereiteten mitochondrialen Fraktionen. Diese Arbeiter fanden 5S in hochgereinigten Mitochondrien und Mitoplasten (Mitochondrien ohne äußere Membran). Schlussfolgerung: 5S rRNA wird in Mitochondrien importiert, aber seine Funktion ist ungewiss.
Visualisierung der mitochondrialen DNA
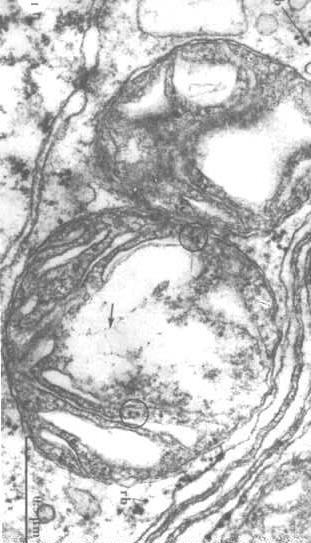
Um die -Struktur der mitochondrialen DNA zu visualisieren, müssen wir die Proteine in der Matrix extrahieren und die DNA aufdecken (Pfeile in der Abbildung rechts).
-Struktur der mitochondrialen DNA zu visualisieren, müssen wir die Proteine in der Matrix extrahieren und die DNA aufdecken (Pfeile in der Abbildung rechts).
Man kann auch Ribosomen in den Kreisen sehen.
Alternativ kann man die DNA extrahieren und auf einer Wasseroberfläche schweben lassen. Dann kann es von einem kunststoffbeschichteten Gitter aufgenommen und im Elektronenmikroskop untersucht werden. Mitochondriale zirkuläre DNA ist in der folgenden Abbildung dargestellt. Diese elektronenmikroskopische Aufnahme stammt aus Fawcett, A Textbook of Histology, Chapman and Hall, 12th edition, 1994.
Mitochondriale Vererbung
Bei Säugetieren wird 99,99% der mitochondrialen DNA (mtDNA) von der Mutter vererbt. Dies liegt daran, dass das Sperma seine Mitochondrien um einen Teil seines Schwanzes trägt und nur etwa 100 Mitochondrien im Vergleich zu 100.000 in der Eizelle hat. Wenn sich die Zellen entwickeln, wird immer mehr mtDNA von Männern verdünnt. Daher ist weniger als ein Teil von 104 oder 0,01% der mtDNA väterlicherseits. Dies bedeutet, dass Mutationen von mtDNA von der Mutter auf das Kind übertragen werden können. Es hat auch Auswirkungen, wenn man Säugetiere mit somatischen Zellen klont. Die Kern-DNA würde von der Spenderzelle stammen, die mtDNA jedoch von der Wirtszelle. So wurde Dolly, das Schaf, geklont.
Es gibt einen Hefestamm namens „Petite“, der strukturell abnormale Mitochondrien aufweist, die nicht oxidativ phosphoryliert werden können. Diese Mitochondrien haben einen Teil oder die gesamte DNA verloren. Mitochondriale Vererbung von Hefe ist biparental, und beide Elternzellen tragen zu den Tochterzellen bei, wenn die haploiden Zellen verschmelzen. Nach Meiose und Mitose erfolgt eine zufällige Verteilung der Mitochondrien auf Tochterzellen. Wenn die Fusion mit Hefe erfolgt, die zierlich ist, und Hefe, die dies nicht ist, ist ein bestimmter Prozentsatz der Tochterzellen „zierlich“.
Mutationen in Säugetier-mtDNA verursachen Krankheiten, weil es eine so kurze Sequenz und einen sehr hohen Informationsgehalt in der Sequenz gibt. Der nächste Dozent zu Mitochondrien in dieser Reihe wird viel Zeit mit dem mitochondrialen Genom verbringen. Da jede Zelle Hunderte von Mitochondrien und Tausende von Kopien des Genoms enthält, können die Auswirkungen der mutierten Mitochondrien verwässert werden. Wie erwartet, sind die Gewebe oder Organe, die am wahrscheinlichsten betroffen sind, am stärksten von der oxidativen Phosphorylierung (ATP-Produktion) abhängig. Bei jungen Menschen kann es nicht abgeholt werden, weil sogar eine Person mit 15% normalen Mitochondrien genug haben könnte, um gesund zu sein. Alternde Patienten können jedoch einen schwereren Krankheitsphänotyp aufweisen.
Einige Beispiele für Krankheiten:
- Leber-erbliche Optikusneuropathie (Degeneration des Sehnervs, begleitet von zunehmender Blindheit): verursacht durch Mutation des Gens, das für die Untereinheit 4 der NADH-C0Q-Reduktase kodiert.
- „zerlumpte Muskelfasern“, die mit ruckartigen Bewegungen verbunden sind, werden durch Mutation der mitochondrialen Lysin-tRNA verursacht.
- Kaerns-Sayre-Syndrom: Augenfehler, abnormaler Herzschlag, Degeneration des Zentralnervensystems. Mehrere große Deletionen in mtDNA.
Kann beschädigte mitochondriale DNA repariert werden?
Aktuelle Studien sagen ja.
- Meeusen, S, Tieu, Q, Wong E, Weiss, E, Schieltz, D, Yates, JR und Nunnari, J. Mgm101p ist eine neuartige Komponente des mitochondrialen Nukleoids, das DNA bindet und für die Reparatur von oxidativ geschädigter mitochondrialer DNA benötigt wird. J Cell Biol 145: 291-304 (1999)
- Mgm steht für „mitochondrial genome maintenance“. Es wurde in Hefezellen bei der Suche nach Mutanten entdeckt, die einen temperaturempfindlichen Verlust von mitochondrialer DNA verursachten.
- Fusionierte Mgm101 mit grün fluoreszierendem Protein und fand heraus, dass es an den punktförmigen „Nukleoid“ -Strukturen lokalisiert war. Die Lokalisierung überlappte sich mit der von DNA-Nachweissystemen.
- Nachdem das Proteinscreening das Mgm101 gefunden hatte, untersuchten sie, wie sich sein Verlust auf die Atmungskompetenz auswirkte. Offensichtlich wurde das Protein für die Funktion benötigt, aber sie wissen nicht genau, welche Rolle es zu diesem Zeitpunkt spielt.
- Betrachtete die COOH-Terminalregion und sah, dass sie sehr einfach war. Das deutet darauf hin, dass das Mgm101p die Fähigkeit haben könnte, DNA zu binden. Verglich seine Bindung an DNA-Cellulosesäulen (unter Hochsalzbedingungen) mit einem anderen bekannten DNA-Bindungsprotein und bestätigte eine relativ hohe Affinitätsbindung durch beide Proteine.
Was passiert mit alten, abgenutzten Mitochondrien?
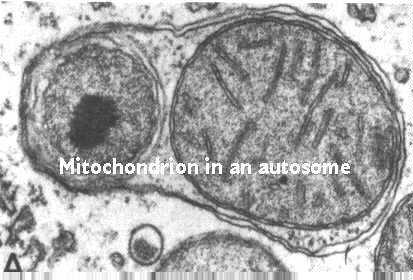
Mitochondriale Zahlen werden durch Autophagie gesteuert. Dies ist ein Prozess, bei dem Lysosomen an der Kontrolle von Zellbestandteilen beteiligt sind. Diese Abbildung zeigt den Prozess; Es ist aus Fawcett, A Textbook of Histology, Chapman und Hall, 12. Auflage, 1994 entnommen.
Der Prozess beginnt, indem endoplasmatische Retikulummembranen um das Mitochondrium gewickelt werden. Dann kommen Vesikel aus dem Golgi-Komplex und verbinden sich mit der autophagischen Vakuole. Diese Vesikel enthalten Hydrolasen, die an die Mannose-6-Phosphatrezeptoren in den Vesikelmembranen gebunden sind. Die Lysosomen-Webseite diskutiert ihre Funktion und ihr Schicksal. Denken Sie daran, dass sie mit der autophagischen Vakuole verschmelzen. Der saure pH-Wert ermöglicht es dann, die Hydrolasen von ihren Rezeptoren zu entfernen. Die Rezeptoren werden in anderen Vesikeln wieder in den Golgi-Komplex zurückgeführt.
In der Zwischenzeit bildet sich das Lysosom, wenn der pH-Wert sinkt und die Zellen beginnen, den Inhalt abzubauen. Denken Sie daran, dass Lysosomen LAMP + sind, aber sie tragen das MPR nicht, da diese in den Golgi-Komplex recycelt wurden. Welcher Mantel befindet sich um die Transportvesikel, die zur autophagischen Vakuole gehen?
Für weitere Informationen wenden Sie sich an:
Gwen Childs, Ph.D.,FAAA
Professor und Lehrstuhl
Abteilung für Neurobiologie und Entwicklungswissenschaften
Universität von Arkansas für medizinische Wissenschaften
Little Rock, AR 72205
Bei Fragen wenden Sie sich bitte an diese E-Mail-Adresse: