
¿Cómo se Replican los Mitocondios?
La replicación mitocondrial está diagramada en la caricatura de la barra lateral y se muestra arriba en una micrografía de electrones. Las mitocondrias se replican como las células bacterianas. Cuando se hacen demasiado grandes, se someten a fisión. Esto implica un surco de la membrana interna y luego de la externa como si alguien estuviera pellizcando la mitocondria. Luego las dos mitocondrias hijas se separaron. Por supuesto, las mitocondrias primero deben replicar su ADN. Esto se discutirá con más detalle en la siguiente sección. Una micrografía electrónica que representa el proceso de surco se muestra en estas figuras. La figura de arriba fue tomada de Fawcett, Un libro de texto de Histología, Chapman y Hall, 12a edición, 1994

A veces, las mitocondrias nuevas se sintetizan de novo en centros ricos en proteínas y polirribosomas necesarios para su síntesis. La micrografía de electrones en la figura anterior muestra un centro de este tipo. Parece que el grupo de mitocondrias está sentado en una matriz de proteínas y otros materiales necesarios para su producción. ¿Cómo podría probar que el material en esa región estaba produciendo proteínas mitocondriales?
Se necesitan ciertas proteínas mitocondriales antes de que las mitocondrias puedan dividirse.
Esto se ha demostrado en un estudio realizado por Sorgo y Yaffe, J Cell Bio. 126: 1361-1373, 1994. Mostraron el resultado de la eliminación de una proteína de la membrana externa de las mitocondrias llamada MDM10. Esta figura muestra los resultados. Las mitocondrias son capaces de absorber componentes y producir membranas y enzimas de matriz. Sin embargo, la fisión no está permitida. Por lo tanto, el resultado es una mitocondria gigante. Esto está mal en la micrografía de abajo.

el ADN Mitocondrial y su función.
Las mitocondrias tienen parte de su propio ADN, ribosomas, y pueden producir muchas de sus propias proteínas. El ADN es circular y se encuentra en el matrix.in estructuras puntiagudas llamadas «nucleoides». Cada nucleoide puede contener de 4 a 5 copias del ADN mitocondrial (ARNm).
El ADN mitocondrial humano es de 16,569 bp; codifica un número de proteínas mitocondriales
- Subunidades 1, 2 y 3 de la citocromo oxidasa
- Subunidades 6, 8,9 de la Fo ATPasa
- Subunidad apocitocromo b de la CoQH2-Citocromo C reductasa
- Siete subunidades de NADH-COQ reductasa
El núcleo codifica las proteínas restantes. La mayor parte de los lípidos se importan (recordemos las conferencias sobre la adición de lípidos a las membranas). Esta caricatura de su texto muestra la participación nuclear. Las etiquetas resaltadas son medicamentos que se pueden usar para bloquear el proceso y probar la fuente de la proteína mitocondrial.

Las mitocondrias también tienen sus propios ribosomas y ARNt:
- 22 ARNt
- ARNr
- 16S
- 12S
- 5S
(Magalhaes, PJ; Andreu, AL, Schon EA, Evidencia de la presencia de ARNr 5 S en Células Mol Biol de mitocondrias de mamíferos 9: 2375-2382)
La Figura de la izquierda muestra los ribosomas mitocondriales como gránulos en las mitocondrias.
Los textos todavía dicen que las mitocondrias no tienen ARNr 5S, sin embargo, el estudio reciente citado anteriormente muestra evidencia de 5S en fracciones mitocondriales cuidadosamente preparadas. Estos trabajadores encontraron 5S en mitocondrias y mitoplastos altamente purificados (mitocondrias sin la membrana externa). Conclusión: El ARNr 5S se importa a las mitocondrias, pero su función es incierta.
Visualización de ADN mitocondrial
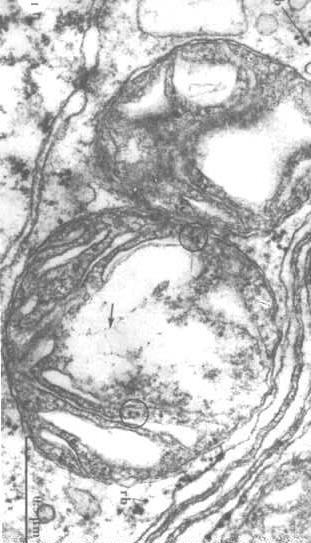
Para visualizar el estructura del ADN mitocondrial, tenemos que extraer las proteínas de la matriz y revelar el ADN (las flechas en la figura a la derecha).
estructura del ADN mitocondrial, tenemos que extraer las proteínas de la matriz y revelar el ADN (las flechas en la figura a la derecha).
También se pueden ver ribosomas en los círculos.
como alternativa, se puede extraer el ADN y flotar en la superficie del agua. Luego, puede ser recogido por una rejilla recubierta de plástico y examinado en el microscopio electrónico. El ADN circular mitocondrial se muestra en la siguiente figura. Esta micrografía electrónica está tomada de Fawcett, Un libro de texto de Histología, Chapman y Hall, 12a edición, 1994.
Herencia mitocondrial
En los mamíferos, el 99,99% del ADN mitocondrial (ADNmt) se hereda de la madre. Esto se debe a que el esperma transporta sus mitocondrias alrededor de una porción de su cola y tiene solo alrededor de 100 mitocondrias en comparación con 100,000 en el ovocito. A medida que las células se desarrollan, más y más del ADNmt de los machos se diluye. Por lo tanto, menos de una parte de cada 104 o 0,01% del ADNmt es paterno. Esto significa que las mutaciones del ADNmt se pueden transmitir de madre a hijo. También tiene implicaciones si se hace clonación de mamíferos con el uso de células somáticas. El ADN nuclear sería de la célula donante, pero el ADNmt sería de la célula huésped. Así es como la oveja Dolly fue clonada.
Hay una cepa de levadura, llamada «Petite» que tiene mitocondrias estructuralmente anormales que son incapaces de fosforilación oxidativa. Estas mitocondrias han perdido parte o todo su ADN. La herencia mitocondrial de la levadura es biparental, y ambas células progenitoras contribuyen a las células hijas cuando las células haploides se fusionan. Después de la meiosis y la mitosis, hay una distribución aleatoria de las mitocondrias a las células hijas. Si la fusión es con levadura que es pequeña y levadura que no lo es, un cierto porcentaje de las células hijas serán «pequeñas».
Las mutaciones en el ADNmt de los mamíferos causan enfermedades, porque hay una secuencia tan corta y un contenido de información muy pesado en la secuencia. El próximo profesor de mitocondrias de esta serie dedicará una gran cantidad de tiempo al genoma mitocondrial. Dado que cada célula contiene cientos de mitocondrias y miles de copias del genoma, los efectos de las mitocondrias mutadas pueden diluirse. Como era de esperar, los tejidos u órganos más propensos a verse afectados serían los más dependientes de la fosforilación oxidativa (producción de ATP). En los jóvenes, es posible que no se capte porque incluso una persona con un 15% de mitocondrias normales podría tener suficiente para estar saludable. Sin embargo, los pacientes de edad avanzada pueden mostrar un fenotipo de enfermedad más grave.
Algunos ejemplos de enfermedades:
- Neuropatía óptica hereditaria de Leber (degeneración del nervio óptico, acompañada de ceguera creciente): causada por mutación en el gen que codifica la subunidad 4 de la NADH-C0Q reductasa.
- las» fibras musculares irregulares » asociadas con movimientos irregulares son causadas por la mutación del ARNt de lisina mitocondrial.
- Síndrome de Kaerns-Sayre: defectos oculares, latidos cardíacos anormales, degeneración del sistema nervioso central. Varias eliminaciones grandes en el ADNmt.
¿Se puede reparar el ADN mitocondrial dañado?
Los estudios actuales dicen que sí.
- Meeusen, S, Tieu, Q, Wong E, Weiss, E, Schieltz, D, Yates, JR, y Nunnari, J. Mgm101p es un nuevo componente del nucleoide mitocondrial que se une al ADN y es necesario para la reparación del ADN mitocondrial dañado oxidativamente. Biol de Células J 145: 291-304 (1999)
- Mgm significa «mantenimiento del genoma mitocondrial». Se descubrió en células de levadura mientras buscaba mutantes que causaban una pérdida sensible a la temperatura del ADN mitocondrial.
- Fusionó Mgm101 a proteína fluorescente verde y encontró que estaba localizada en las estructuras «nucleoides» puntiagudas. Localización superpuesta con la de los sistemas de detección de ADN.
- Después de que el cribado de proteínas encontró el Mgm101, estudiaron cómo su pérdida afectó la competencia respiratoria. Es evidente que la proteína era necesaria para funcionar, pero no saben exactamente cuál es su papel en este momento.
- Miró la región terminal de COOH y vio que era muy básica. Eso sugirió que el Mgm101p podría tener la capacidad de unirse al ADN. Se comparó su unión a columnas de celulosa de ADN (en condiciones de alta salinidad) con otra proteína de unión al ADN conocida y se confirmó una unión de afinidad relativamente alta por ambas proteínas.
¿Qué sucede con las mitocondrias viejas y desgastadas?
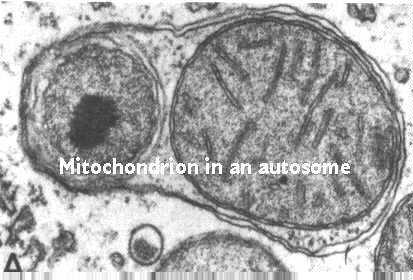
Los números mitocondriales se controlan por autofagia. Este es un proceso por el cual los lisosomas participan en el control de los constituyentes celulares. Esta figura muestra el proceso; está tomada de Fawcett, A Textbook of Histology, Chapman and Hall, 12ª edición, 1994.
El proceso comienza envolviendo las membranas del retículo endoplásmico alrededor de la mitocondria. Luego, las vesículas provienen del complejo de Golgi y se unen a la vacuola autofágica. Estas vesículas contienen hidrolasas unidas a los receptores de fosfato de manosa 6 en las membranas de las vesículas. La página web de lisosomas discute su función y destino. Recordemos que se fusionan con la vacuola autofágica. El pH ácido permite que las hidrolasas se eliminen de sus receptores. Los receptores se reciclan de nuevo al complejo de Golgi en otras vesículas.
Mientras tanto, el lisosoma se forma a medida que el pH disminuye y las células comienzan a degradar el contenido. Recuerde que los lisosomas son LAMP+, pero no llevan el MPR porque han sido reciclados en el Complejo Golgi. ¿Qué capa se encuentra alrededor de las vesículas de transporte que van a la vacuola autofágica?
Para más información, póngase en contacto con:
Gwen Childs, Ph. D., FAAA
Profesor y Presidente
Departamento de Neurobiología y Ciencias del Desarrollo
Universidad de Arkansas para Ciencias Médicas
Little Rock, AR 72205
Para preguntas, póngase en contacto con esta dirección de correo electrónico: