Úvod
Infantilní neuroaxonální dystrofie (INAD) je autosomálně recesivní, vzácné neurodegenerativní onemocnění neznámé frekvenci. Nástup příznaků se obvykle vyskytuje mezi 6 měsíci a 3 lety věku. Před touto dobou se děti vyvíjejí normálně. Prvním příznakem INAD může být zpomalení dosažení normálních vývojových milníků nebo regrese ve vývojových milnících (Ramanadham et al., 2015). Kmen hypotonie, strabismus a nystagmus jsou časné příznaky onemocnění (Gregory et al ., 2017). Progrese onemocnění je rychlá a jak postupuje, ztrácí se více získaných dovedností. Svaly se brzy stanou hypotonickými a později se stanou spastickými (Levi a Finazzi, 2014). Nakonec se ztratí veškerá dobrovolná kontrola svalů. Svalová slabost může také vést k potížím s krmením a dýcháním. Kromě nystagmu dochází u některých dětí ke ztrátě zraku. Kognitivní funkce se postupně ztrácejí a rozvíjí se demence. Životnost je obvykle 5 až 10 let (Gregory et al., 1993; Jardim et al., 2004; Macauley and Sands, 2009).
Molekulární Patologie
Infantilní Neuroaxonální Dystrofie patří do skupiny neurodegenerativních poruch, které zahrnuje atypické pozdním nástupem neuroaxonální dystrofie (PLÁNOVANÝ) a dystonie, Parkinsonismus komplex (DPC). Většina případů INAD je spojena s homozygotními nebo složenými heterozygotními mutacemi v genu PLA2G6, které ovlivňují katalytickou aktivitu jeho proteinového produktu (Engel et al ., 2010). Na PLA2G6 gen kóduje skupinu prostřednictvím vápník-nezávislé fosfolipázy A2 protein (PLA2G6 nebo iPLA2ß, ∼85/88 kDa) s lipázy a sedm ankyrin repeat-obsahující domény (Tang et al., 1997). PLA2G6 hydrolyzuje SN-2 acylový řetězec fosfolipidů a vytváří volné mastné kyseliny a lysofosfolipidy.
Fosfolipidů ve vnitřní membráně mitochondrií jsou bohaté na nenasycené mastné kyseliny v sn-2 pozici, zejména kardiolipin (Seleznev et al., 2006). Tyto nenasycené mastné kyseliny jsou zvláště náchylné k hojné reaktivní formy kyslíku produkované v mitochondriích (Murphy, 2009), což v peroxidized fosfolipidů ve vnitřní membráně mitochondrií. PLA2G6 lokalizuje do mitochondrií (Williams a Gottlieb, 2002; Liou et al., 2005) v souladu se zvýšenou poptávkou po hydrolýze peroxidized mastných kyselin v sn-2 poloze fosfolipidů vede k přestavěný fosfolipidů (Balsinde et al., 1995; Zhao a kol., 2010). Když je PLA2G6 vadný, je poškozena integrita vnitřní membrány mitochondrií. PLA2G6 se také lokalizuje na axon (Ong et al., 2005; Seleznev et al., 2006), což svědčí o zvýšené lokalizované poptávce po remodelaci fosfolipidů. Projevem takové akumulace v mozku je jedinečný na klíčové oblasti mozku, jako jsou bazální ganglia, která vyústila v různé názvy pro stejnou základní molekulární patogeneze zahrnující PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
Ultrastrukturní analýza neuronů u KNOCKOUTOVÝCH myší PLA2G6 je v souladu s touto molekulární patologií. Mitochondrie s větvení a tubulární cristae, mitochondrie s degenerované cristae, axony s cytoskeletu kolaps, a částečné membrány, ztráta axonů bylo pozorováno (Beck et al., 2011). Na mikroskopické úrovni se tyto rysy objevují jako axonální otoky a sféroidní těla v pre-synaptických terminálech (Obrázek 1) v centrálním nebo periferním nervovém systému.
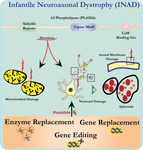
Obrázek 1. Infantilní neuroaxonální dystrofie (INAD) je neurodegenerativní porucha související s mutacemi v genu PLA2G6. Různé mutace PLA2G6 vedou k dysfunkční A2 fosfolipáze, která vede k defektům mitochondriální a axonální membrány. Tyto defekty způsobují poškození neuronů vizualizované jako axonální otoky a akumulaci pre-synaptických sféroidů. Náhrada enzymů za účelem obnovení funkcí, náhrada genů nebo úprava pro korekci defektního PLA2G6 jsou navrženy terapeutické strategie.
Molekulární Diagnostika a Vzácné Onemocnění, Posílení postavení Pacienta
na Rozdíl od specifických klinických, elektrofyziologických a zobrazovacích funkcí, před dostupnost další generace sekvenování, kožní biopsie ukazuje axonální otoky a hyperboloidu subjektů v pre-synaptické terminály v centrální nebo periferní nervový systém jsou diagnostická kritéria pro potvrzení INAD (Gregory et al., 2017; Jodice a kol., 2017). K potvrzení diagnózy bylo často vyžadováno více biopsií. Rodiny obecně čekaly na diagnózu mnoho let. S klesající náklady genů a sekvenování genomu, dostupnost cílené gen panel testování diagnostické laboratoře pro diagnostikováno neurologické onemocnění, a zvýšit povědomí lékařů o dostupnosti genetická diagnostika, rodiny dostávají diagnózu rychleji, někdy v roce prvním příznakem se objeví. Děti v těchto rodinách jsou stále mladé a rodiny jsou motivovány k partnerství s vědci, aby našli léčbu nemoci svých dětí. Za účelem financování výzkumného procesu vytvořila skupina rodičů pacientů s INAD Nadaci INADcure. Nadace vyvolalo značné finanční prostředky na výzkum a spolupracuje s Vzácné Genomics Institute, aby jim průvodce v udělování výzkumných grantů. Zajímavé je, že genetická analýza 22 indických rodin z roku 2016 s INAD, ANAD a DPC zjistila, že 10/22 rodin (45,45%) postrádalo mutace v oblasti kódování genu PLA2G6 (Kapoor et al ., 2016). Selhání identifikace mutací v kódující oblasti PLA2G6 zdůrazňuje, že budoucnost molekulární diagnostiky úsilí by vyžadovalo celý gen sekvenování k identifikaci mutace v intronic a regulačních oblastí z PLA2G6 genu v INAD postižených pacientů. Dále v případech, kdy nejsou pozorovány mutace spojené s PLA2G6, naznačuje, že příčina onemocnění může být způsobena mutací v jiných genech než PLA2G, které je třeba prozkoumat.
Možné Terapie
Většina terapií, které jsou považovány za vzácné onemocnění, jako INAD zahrnující vadný enzym je enzym, výměna, nahrazování genů nebo genových korekce. Pokud je nedostatek enzymu způsoben recesivní genetickou vadou, předpokládá se, že nahrazení nebo doplnění enzymu může problém napravit (Smith et al., 2012; Yu-Wai-Man, 2016). Pokusy, které mají prokázat, že terapie poskytující správný gen nebo enzym zachrání fenotyp INAD, se však teprve provádějí a testují.
enzymová substituční terapie (ERT)
vzhledem k tomu, že mozek je primárním orgánem postiženým INAD, enzymová substituční terapie INAD by s největší pravděpodobností vyžadovala infuzi enzymu do mozku. V roce 2017, Biomarin obdržel schválení FDA pro tripeptidyl peptidázy 1 (cerliponase alfa) jako léčba základní příčiny Batten nemoc, deficit tripeptidyl peptidázy (TPP1), lysosomálního enzymu (US Food Drug Administration, 2017). Tripeptidyl peptidáza 1 (cerliponáza alfa; ∼59 kDa) je první ERT, která se přímo podává do mozkomíšního moku (CSF) mozku. Jiné léky ERT, které mohou být podávány do CSF, jsou v klinických studiích (Jardim et al ., 2004; Macauley and Sands, 2009).
účinnost tripeptidyl peptidázy 1 (cerliponase alfa) na schopnost chůze Batten onemocnění pacientů ukazuje, že enzym substituční terapie pro onemocnění, které mají vliv na mozek je to teoreticky možné. Cílení nahrazení enzymu do mitochondrií pro INAD bude náročnější než lysosomální cílení, které již byly úspěšné IV ERT na dlouhou dobu, jako v případě Gaucherovy choroby. Proto, tam je ještě mnoho biologických otázek specifických pro INAD, které je třeba vyřešit:
• Dělá terapie pro INAD třeba mechanismy, které mohou případně doprava PLA2G6 do buňky?
* jaký mechanismus lze použít k případné lokalizaci PLA2G6 do mitochondrií?
• je to dostatečné nebo bude PLA2G6 také nutné lokalizovat do axonu a pokud ano, jak?
* existuje zvláštní potřeba řešit cílení periferního nervového systému?
* může částečný PLA2G6 zachránit svůj nedostatek nebo je vyžadován celý protein?
* jaké je vhodné rozpouštědlo pro dodávání genového produktu PLA2G6 do mozkomíšního moku?
• pokud enzym PLA2G6 není 100% čistý, jaké jsou jeho kontaminanty a jsou bezpečné?
• Budou nějaké generalizované intravaskulární komplikace vzhledem k PLA2G6 enzymu infuzi do CNS? Má infuzní dávka nějaký význam pro takové komplikace?
dalším problémem je, že enzymatická substituční terapie vyžaduje počáteční umístění intracerebroventrikulárního katétru a časté infuze. Umístění katétru vyžaduje anestezii a infuze mohou vyžadovat anestezii v závislosti na spolupráci pacientů. Řešení obav pediatrických neurologů a rodičů dětí s INAD s ohledem na použití anestezie u pacientů s INAD s příslušnými informacemi bude vyžadovat další úsilí.
Genové Terapie/Gen Náhradní
lidské PLA2G gen je kolem 70 kb s 17 exonů a 2 alternativní exons. Nejdelší transkript kódující proteiny je však pouze 3,3 kb a sekvence kódující proteiny je něco přes 2,4 kb, které lze snadno zabalit do virového vektorového nákladu. Budeme nejprve prozkoumat další lysozomální střádání poruchy v CNS, které mají předklinické údaje se pak porovnají s INAD. Nedostatek preklinických dat ERT v INAD je významnou mezerou ve znalostech v oboru,a proto je zásadní, abychom pozorovali další preklinické modely poruch CNS, které procházejí studiemi genové terapie. Dříve, preklinické studie na myších určeno pro genovou terapii pro lysosomální porucha ukládání mucopolysaccharidoses typ IIIA (MPS-IIIA) balení správnou verzi sulphamidase gen uvnitř virové vektory (Sorrentino et al., 2013). Studie využila rostoucí porozumění hematoencefalickou bariérou (BBB), které aktivně reguluje transport velkých molekul z krve do CNS pomocí procesu zvaného transcytosis (Pardridge, 2005b; Sorrentino a Fraldi, 2016).
Transkytóza zahrnuje endocytózu ligand na luminální straně, zprostředkovanou receptory specifickými pro ligand (např.,) obohacený o kapilární endotel (Pardridge, 2005a). Následuje pohyb endocytovaného nákladu endotelovou cytoplazmou a nakonec exocytóza na abluminální (mozkové) straně, čímž se účinně doručuje náklad přes BBB (Pardridge, 2002). MPS-IIIA preklinické studie vyrobeny chimérická sulphamidase protein, který obsahoval BBB-vazebnou doménu od apolipoproteinu B k usnadnění vychytávání endotelu a také signálního peptidu z iduronát-2-sulfatázy na podporu efektivní exocytóza k abluminal straně BBB (Sorrentino et al., 2013). Virový vektor nákladní kódování chimérická sulphamidase byl pak vložen do adeno-asociovaný virus (AAV) sérotyp 2/8 cílení jater (Sorrentino a Fraldi, 2016). Tak játra sloužil jako interní továrna, která poskytovala stálý přísun chimerická-sulphamidase, což vedlo k 10-15% zvýšení v mozku sulphamidase činnost i 7 měsíců po jaterní genovou terapii. Toto zvýšení hladin aktivity sulfamidázy v mozku vedlo k kvantifikovatelnému zlepšení patologie mozku a výsledků chování v myším modelu MPS-IIIA (Sorrentino et al ., 2013).
pokud by byla podobná studie provedena pro INAD, odpověděla by na několik kritických otázek týkajících se náhrady enzymů za terapii INAD. Alternativně může intra-vaskulární nebo intra-CSF podávání produktů genové terapie na bázi aav9 přímo cílit na CNS (Bey et al., 2017; Roca a kol., 2017). Mnohočetné mutace, které mají pacienti s INAD na svém genu PLAG26, však poskytují jedinečné výzvy genové terapii. Ačkoli velikost genu PLA2G6 2-3 kb by neměla představovat problém pro jeho vložení do virového vektoru, regulační komplikace z korekce PLA2G6 je obtížné předvídat. Regulační komplikace se stávají nekontrolovatelnými, zejména pokud genová terapie nemůže být správně lokalizována v cílových tkáních. Enzymatická substituční terapie může být také problémem, pokud se u některých mutantních produktů ukáže, že jsou dominantní negativní na plag26 divokého typu.
většina výzev možných terapií pro INAD pochází ze skutečnosti, že se jedná o velmi vzácné onemocnění. Skutečnost, že INAD má známou genetickou etiologii, však poskytuje cesty pro možné terapie. Každá úspěšná léčba INAD navíc obdrží status léčivého přípravku pro vzácná onemocnění a veškerou ochranu, kterou získá, protože INAD je vzácné onemocnění. Tak i přes všechny výše uvedené problémy, orphan drug status poskytuje silnou motivaci pro vzácná onemocnění, vědci a biotechnologického průmyslu, nemluvě o tom, když se genetická příčina je již známa.
editace genu / báze
od roku 2017 bylo v lidském genu PLA2G6 pozorováno nejméně 277 mutací missense (Lek et al ., 2016). Pouze malá část mutací PLA2G6 zahrnuje posuny rámců, indely, nesmyslné mutace a mutace ve spojovacích místech (Morgan et al ., 2006). Korekce genu v populaci cílových buněk nebo korekce mutovaných dna bází je tedy atraktivní terapeutickou cestou. V průběhu let bylo vyvinuto několik nástrojů pro úpravy gen, a CRISPR/Cas9-based editace genomu je oslavován jako průlomovou technologií s klinickou studií plánováno v roce 2018. Tyto technologie využívají DNA vázající protein, který může také štěpit vlákno stanoveným způsobem, aby se prostor pro vložení nové sekvence DNA nebo korekce škodlivé bází DNA (LaFountaine et al., 2015). Technologie CRISPR / Cas9 již přinesla ohromující terapeutické výsledky v několika předklinických modelech onemocnění (např.,) (Dai et al., 2016). Navíc, rostoucí porozumění lidské variace představuje novější výzvy pro genovou terapii a řízení inovací směrem k opravdové personalizaci gen editaci (Lessard et al., 2017; Scott a Zhang, 2017). Nové technologie, jako je vslendr, virus AAV a technologie zprostředkovaná CRISPR/Cas9, které nahrazují vadné geny v neuronech a dalších buňkách nervového systému, tlačí technologie úpravy genů na nové hranice (Nishiyama et al., 2017).
závěr
infantilní Neuroaxonální dystrofie je závažné neurodegenerativní onemocnění s určitou morbiditou a mortalitou. Toto vzácné onemocnění nabízí vzrušující příležitost k revalidaci dostupných způsobů terapie nové generace a generování novějších. Očekává se, že vznikající shoda v klinických diagnostických kritériích pro INAD poskytne podnět k lepší molekulární diagnostice. Očekává se, že tento pokrok nás povede k vývoji cenově dostupných terapií, které by poskytly kvantifikovatelné zlepšení kvality života pacientů s INAD a zpomalily nebo zlepšily progresi onemocnění. Zdůraznili jsme některé příběhy o úspěchu v Batten nemoc a mucopolysaccharidoses, které nám poskytují inspiraci, klást správné otázky, aby se INAD terapie realitou. Kromě toho by rostoucí virové a nevirové přístupy k úpravě genů založené na CRISPR / Cas9 měly také otevřít novější terapeutické cesty pro INAD.
autorské příspěvky
PB a FA připravily předběžný návrh. SR A AC upravily a přidaly do rukopisu další sekce. DF, AP a LP upravily rukopis.
Prohlášení o střetu zájmů
autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
Balsinde, J., Bianco, I. d., Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. a. (1995). Inhibice fosfolipázy A2 nezávislé na vápníku zabraňuje inkorporaci kyseliny arachidonové a remodelaci fosfolipidů v MAKROFÁGECH P388D1. Proc. Natle. Acad. Věda. USA 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Efektivní cílení na CNS u dospělých myší intratekální infuzí jednovláknové AAV9-GFP pro genovou terapii neurologických poruch. Gene There. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 pro genovou terapii in vivo: slib a překážky. Molo. Další. Nukleové kyseliny 5:e349. doi: 10.1038 / mtna.2016.58
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Engel, L. A., Jing, Z., O ‚ Brien, D. E., Sun, M., and Kotzbauer, P. T. (2010). Katalytická funkce PLA2G6 je narušena mutacemi spojenými s infantilní neuroaxonální dystrofií, ale nikoli dystonií-parkinsonismem. PLoS One 5: e12897. doi: 10.1371 / deník.pone.0012897
PubMed Abstraktní | CrossRef celý Text
Řehoř, A., Kuriane, M. a., Maher, E. R., Hogarth, P., a Hayflick, S. J. (1993). v PLA2G6-Spojené Neurodegeneraci, eds M. P. Adam, h. H. Ardinger, a. R. a. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Řehoř, A., Kuriane, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Zapojení CNS do Fabryho choroby: klinické a zobrazovací studie před a po 12 měsících enzymatické substituční terapie. J. Metab. Dis. 27, 229–240. doi: 10.1023/B: BOLI.0000028794.04349.91
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Spíše M. I., Bhat, V., Gopinath, S., et al. (2016). Genetická analýza PLA2G6 ve 22 indických rodinách s infantilní neuroaxonální dystrofií, atypickou neuroaxonální dystrofií s pozdním nástupem a komplexem parkinsonismu dystonie. PLoS One 11: e0155605. doi: 10.1371 / deník.pone.0155605
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
LaFountaine, J. S., Fathe, K., a Smyth, H. D. (2015). Dodávka a terapeutické aplikace genových editačních technologií ZFNs. TALENs, a CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Lek, m., Karczewski, k., Minikel, e., Samocha, K. E., Banks, e., Fennell, T., et al. (2016). Analýza genetických variací kódujících proteiny u 60 706 lidí. Příroda 536, 285-291. doi: 10.1038/nature19057
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Lidská genetická variace mění CRISPR-Cas9 on-a off-cílení specificita na terapeuticky zapletených lokusech. Proc. Natle. Acad. Věda. U. S. A. 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Levi, s., and Finazzi, d. (2014). Neurodegenerace s akumulací železa v mozku: aktualizace patogenních mechanismů. Před. Farmakol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Plný Text | Google Scholar
Liou, J. Y., Aleksič, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Mitochondriální lokalizace cyklooxygenázy-2 a fosfolipázy A2 nezávislé na vápníku v lidských rakovinných buňkách: důsledky rezistence na apoptózu. Expo. 306, 75-84. doi: 10.1016 / j. yexcr.2005.01.011
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Macauley, S. L., and Sands, MS (2009). Slibná enzymatická substituční terapie zaměřená na CNS u lysozomálních chorob skladování. Expo. Neurol. 218, 5–8. doi: 10.1016 / j. expneurol.2009.03.040
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Mehnaaz, L. (2016). Neurodegenerace s akumulací železa v mozku (NBIA) dříve Hallervorden-Spatzova choroba. J.Doc. Phys. Indie 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K. Morton, J. E. V., Řehoř, A., Gissenovi, P., Sonek, S., et al. (2006). PLA2G6, kódující fosfolipázu A2, je mutován u neurodegenerativních poruch s vysokým obsahem železa v mozku. Adresa. Genete. 38, 752–754. doi: 10.1038/ng1826
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Murphy, M. P. (2009). Jak mitochondrie produkují reaktivní druhy kyslíku. Biochem. J. 417, 1-13. doi: 10.1042/BJ20081386
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C., a Faria, A. (2016). Neurodegenerace s akumulací železa v mozku: kazuistika. Dement. Neuropsychol. 10, 160–164. doi: 10.1590/S1980-5764-2016DN1002014
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Nishiyama, J., Mikuni, T., a Yasuda, R. (2017). Editace genomu zprostředkovaná virem pomocí opravy zaměřené na homologii v mitotických a postmitotických buňkách v mozku savců. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j. neuron.2017.10.004
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, a. S.-F., a Farooqui, a. a. (2005). Distribuce fosfolipázy A2 nezávislé na vápníku (iPLA2) v opičím mozku. J. Neurocytol. 34, 447–458. doi: 10.1007/s11068-006-8730-4
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Pardridge, W. M. (2002). Cílení léků a genů do mozku pomocí molekulárních trojských koní. Adresa. Rev. Drug Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Pardridge, W. m. (2005a). Molekulární biologie hematoencefalické bariéry. Molo. Biotechnol. 30, 57–70. doi: 10.1385 / MB: 30: 1:057
CrossRef Plný Text / Google Scholar
Pardridge, W. m. (2005b). Hematoencefalická bariéra: úzký profil ve vývoji mozkových drog. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Kostí, R. N., Hancock, W. D., Lei, X., et al. (2015). Fosfolipázy A2 nezávislé na vápníku a jejich role v biologických procesech a onemocněních. J. Lipid Res.56, 1643-1668. doi: 10.1194 / MLR.R058701
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, a., Sánchez, v., Sánchez, X., et al. (2017). Korekce onemocnění genovou terapií zprostředkovanou AAV v novém myším modelu mukopolysacharidózy typu IIID. Hučení. Molo. Genete. 26, 1535–1551. doi: 10.1093/hmg/ddx058
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Scott, D. A., a Zhang, F. (2017). Důsledky lidské genetické variace v úpravě terapeutického genomu založené na CRISPR. Adresa. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K., and Ma, Z .a. (2006). Fosfolipáza A2 nezávislá na vápníku lokalizuje a chrání mitochondrie během apoptotické indukce staurosporinem. J.Biol. Cheme. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Smith, a. J., Bainbridge, J. W., a Ali, R. R. (2012). Genová suplementační terapie pro recesivní formy dědičných retinálních dystrofií. Gene There. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Sorrentino, N. C., D ‚ Orsi, L., Sambri, i., Nusco, e., Monako, C., Spampanato, C., et al. (2013). Vysoce vylučovaná sulfamidáza navržená tak, aby procházela hematoencefalickou bariérou, koriguje mozkové léze myší s mukopolysacharidózami typu IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Sorrentino, N. C., and Fraldi, a. (2016). Cílení mozku v MPS-IIIA. Pediatr. Endokrinol. Rev.13(Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., and Jones, S. S. (1997). Nová cytosolická fosfolipáza A2 nezávislá na vápníku obsahuje osm ankyrinových motivů. J.Biol. Cheme. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
US Food and Drug Administration (2017). FDA schvaluje první léčbu formy Battenovy choroby. K dispozici na: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., and Gottlieb, R. a. (2002). Inhibice mitochondriální fosfolipázy A2 nezávislé na vápníku (iPLA2) zmírňuje ztrátu mitochondriálních fosfolipidů a je kardioprotektivní. Biochem. J. 362 (Bod 1), 23-32. doi: 10.1042/bj3620023
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Yu-Wai-Man, P. (2016). Genetická manipulace s dědičnými neurodegenerativními chorobami: mýtus nebo realita? Br. J. Oftalmol. 100:1322. doi: 10.1136/bjophthalmol-2015-308329
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., a kol. (2010). Ochrana beta-buněk pankreatu skupinou prostřednictvím fosfolipázy A (2) zprostředkované opravy peroxidace mitochondriální membrány. Endokrinologie 151, 3038-3048. doi: 10.1210 / en.2010-0016
PubMed Abstrakt / CrossRef Plný Text / Google Scholar