wprowadzenie
infantylna dystrofia neuroaksonalna (INAD) jest autosomalną recesywną rzadką chorobą neurodegeneracyjną o nieznanej częstości występowania. Objawy pojawiają się zwykle między 6 miesiącem a 3 rokiem życia. Przed tym czasem niemowlęta rozwijają się normalnie. Pierwszym objawem INAD może być spowolnienie osiągnięcia normalnych etapów rozwoju lub regresji w etapach rozwoju (Ramanadham et al., 2015). Hipotonia tułowia, zeza i oczopląsu są wczesne objawy choroby (Gregory et al., 2017). Postęp choroby jest szybki, a w miarę jej postępu traci się więcej nabytych umiejętności. Mięśnie szybko stają się hipotoniczne, a później spastyczne (Levi and Finazzi, 2014). Ostatecznie, wszystkie dobrowolne kontroli mięśni traci. Osłabienie mięśni może również prowadzić do trudności w karmieniu i oddychaniu. Oprócz oczopląsu, niektóre dzieci doświadczają utraty wzroku. Funkcje poznawcze są stopniowo tracone i rozwija się demencja. Długość życia wynosi zwykle od 5 do 10 lat (Gregory et al., 1993; Jardim et al., 2004; Macauley and Sands, 2009).
patologia molekularna
infantylna dystrofia Neuroaksonalna należy do rodziny zaburzeń neurodegeneracyjnych, która obejmuje atypową późną dystrofię neuroaksonalną (ANAD) i zespół dystonii parkinsonizmu (DPC). Większość przypadków INAD wiąże się z homozygotycznymi lub heterozygotycznymi mutacjami w genie PLA2G6, które wpływają na aktywność katalityczną jego produktu białkowego (Engel i wsp ., 2010). Gen PLA2G6 koduje grupę poprzez niezależne od wapnia białko fosfolipazy A2 (PLA2G6 lub iPLA2ß, ∼85/88 kDa) z lipazą i siedmioma domenami zawierającymi powtórzenia ankyriny (Tang i in., 1997). PLA2G6 hydrolizuje łańcuch acylowy Sn-2 fosfolipidów, wytwarzając wolne kwasy tłuszczowe i lizofosfolipidy.
fosfolipidy w wewnętrznej błonie mitochondriów są bogate w nienasycone kwasy tłuszczowe w pozycji sn-2, szczególnie kardiolipiny (Seleznev i wsp ., 2006). Te nienasycone kwasy tłuszczowe są szczególnie wrażliwe na obfite reaktywne formy tlenu wytwarzane przez mitochondria (Murphy, 2009), w wyniku czego powstają peroksydowane fosfolipidy w wewnętrznej błonie mitochondriów. PLA2G6 lokalizuje się w mitochondriach (Williams and Gottlieb, 2002; Liou et al., 2005) w związku ze zwiększonym zapotrzebowaniem na hydrolizę peroksydowanych kwasów tłuszczowych w pozycji sn-2 fosfolipidów, co prowadzi do przebudowy fosfolipidów (Balsinde et al., 1995; Zhao et al., 2010). Gdy PLA2G6 jest uszkodzony, wewnętrzna integralność membrany mitochondriów jest uszkodzona. PLA2G6 również lokalizuje się w aksonie (Ong et al., 2005; Seleznev et al., 2006) wskazując na zwiększone lokalne zapotrzebowanie na przebudowę fosfolipidów. Manifestacja takiej akumulacji w mózgu jest unikalna dla kluczowych obszarów mózgu, takich jak zwoje podstawne, które doprowadziły do różnych nazw tej samej podstawowej patogenezy molekularnej z udziałem PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
analiza ultrastruktury neuronów u myszy nokautujących PLA2G6 jest zgodna z tą patologią molekularną. Zaobserwowano Mitochondria z rozgałęzionymi i rurkowymi cristae, mitochondria ze zdegenerowanymi cristae, aksony z zapadnięciem cytoszkieletu i częściową utratą błony na zaciskach aksonu (Beck et al., 2011). Na poziomie mikroskopowym cechy te pojawiają się jako obrzęki aksonalne i ciała sferoidalne w terminalach przedsynaptycznych (ryc. 1) w centralnym lub obwodowym układzie nerwowym.
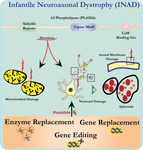
Rysunek 1. Infantylna dystrofia neuroaksonalna (INAD) jest zaburzeniem neurodegeneracyjnym związanym z mutacjami w genie PLA2G6. Różne mutacje PLA2G6 prowadzą do dysfunkcjonalnej fosfolipazy A2, która prowadzi do wad błony mitochondrialnej i aksonalnej. Defekty te powodują uszkodzenia neuronów w postaci obrzęków aksonalnych i gromadzenia się sferoidów przedsynaptycznych. Enzymatyczna wymiana w celu przywrócenia funkcji, wymiana genów lub edycja w celu skorygowania wadliwego PLA2G6 są proponowane strategie terapeutyczne.
diagnostyka molekularna i upodmiotowienie pacjentów z rzadkimi chorobami
oprócz specyficznych cech klinicznych, elektrofizjologicznych i obrazowych, przed dostępnością sekwencjonowania nowej generacji, biopsje skóry wykazujące obrzęki aksonalne i ciała sferoidalne w terminalach przedsynaptycznych w centralnym lub obwodowym układzie nerwowym były kryteriami diagnostycznymi potwierdzenia INAD (Gregory et al., 2017; Iodice et al., 2017). Często, wiele biopsji były wymagane w celu potwierdzenia diagnozy. Rodziny na ogół czekały wiele lat na diagnozę. Wraz ze zmniejszającymi się kosztami sekwencjonowania genów i genomów, dostępnością ukierunkowanych testów paneli genowych z laboratoriami diagnostycznymi dla niezdiagnozowanych chorób neurologicznych oraz rosnącą świadomością lekarzy na temat dostępności diagnostyki genetycznej, rodziny otrzymują diagnozę szybciej; czasami w ciągu roku od pojawienia się pierwszego objawu. Dzieci w tych rodzinach są wciąż młode, a rodziny są zmotywowane do współpracy z naukowcami w celu znalezienia leczenia choroby ich dzieci. W celu sfinansowania procesu badawczego grupa rodziców pacjentów z INAD utworzyła Fundację INADcure. Fundacja zgromadziła znaczne środki na badania i współpracuje z Rare Genomics Institute, aby poprowadzić ich w przyznawaniu grantów badawczych. Co ciekawe, analiza genetyczna 2016 indyjskich rodzin z INAD, ANAD i DPC wykazała, że 10/22 rodzin (45,45%) brakowało mutacji w regionie kodującym Gen PLA2G6 (Kapoor et al., 2016). Brak identyfikacji szkodliwych mutacji w regionie kodującym PLA2G6 podkreśla, że przyszłe badania diagnostyczne molekularne wymagałyby sekwencjonowania całego genu w celu identyfikacji mutacji w regionach intronowych i regulacyjnych genu PLA2G6 u pacjentów dotkniętych INAD. Ponadto przypadki INAD, w których nie obserwuje się mutacji związanych z PLA2G6, sugerują, że przyczyną choroby może być mutacja w genach innych niż PLA2G, która musi zostać zbadana.
możliwe terapie
większość terapii, które są rozważane w przypadku rzadkiej choroby, takiej jak INAD, z udziałem wadliwego enzymu, to wymiana enzymów, wymiana genów lub korekcja genów. Gdy niedobór enzymu jest spowodowany recesywną wadą genetyczną, zakłada się, że wymiana enzymu lub suplementacja może rozwiązać problem (Smith et al., 2012; Yu-Wai-Man, 2016). Jednak eksperymenty mające na celu udowodnienie, że terapie dostarczające prawidłowy gen lub enzym uratują fenotyp INAD, nie zostały jeszcze przeprowadzone i przetestowane.
enzymatyczna terapia zastępcza (ERT)
ponieważ mózg jest głównym organem dotkniętym INAD, enzymatyczna terapia zastępcza dla INAD najprawdopodobniej wymagałaby infuzji enzymu do mózgu. W 2017 r. Biomaryna otrzymała zatwierdzenie FDA dla tripeptydylopeptydazy 1 (cerliponazy alfa) jako leczenia podstawowej przyczyny choroby Battena, niedoboru tripeptydylopeptydazy (TPP1) enzymu lizosomalnego (U. S. Food Drug Administration, 2017). Peptydaza tripeptydylowa 1 (cerliponaza alfa; ∼59 kDa) jest pierwszym ERT podawanym bezpośrednio do płynu mózgowo-rdzeniowego (CSF) mózgu. Inne leki ERT, które można podawać do płynu mózgowo-rdzeniowego, są w badaniach klinicznych (Jardim i wsp ., 2004; Macauley and Sands, 2009).
skuteczność tripeptydylopeptydazy 1 (cerliponazy alfa) na zdolność chodzenia pacjentów z chorobą Battena pokazuje, że enzymatyczna terapia zastępcza w chorobach wpływających na mózg jest teoretycznie możliwa. Celowanie enzymu zastępczego do mitochondriów w przypadku INAD będzie trudniejsze niż celowanie lizosomalne, które już od dłuższego czasu było skuteczne przy dożylnym leczeniu ERT, jak w przypadku choroby Gauchera. Dlatego nadal istnieje wiele problemów biologicznych specyficznych dla INAD, które należy rozwiązać:
• czy terapia INAD wymaga mechanizmów, które mogą ewentualnie transportować PLA2G6 do komórki?
• jaki jest odpowiedni rozpuszczalnik do dostarczania produktu genu PLA2G6 do płynu mózgowo-rdzeniowego?
dodatkowym problemem jest to, że enzymatyczna terapia zastępcza wymaga wstępnego umieszczenia cewnika wewnątrzkomorowego i częstych infuzji. Umieszczenie cewnika wymaga znieczulenia, a napary mogą wymagać znieczulenia w zależności od współpracy pacjentów. Zajęcie się problemami neurologów dziecięcych i rodziców dzieci z INAD w odniesieniu do stosowania znieczulenia u pacjentów z Inad z odpowiednimi informacjami będzie wymagało dalszych wysiłków.
terapia genowa/Wymiana genów
ludzki gen PLA2G ma około 70 kb z 17 eksonami i 2 alternatywnymi eksonami. Najdłuższy transkrypt kodujący białko ma jednak zaledwie 3,3 kb, a Sekwencja kodująca białko ma nieco ponad 2,4 kb, które można łatwo zapakować w ładunek wektora wirusowego. Najpierw zbadamy inne zaburzenia przechowywania lizosomalnego w OUN, które mają dane przedkliniczne, aby następnie porównać je z INAD. Brak danych przedklinicznych ERT w INAD jest istotną luką w wiedzy w tej dziedzinie, dlatego ważne jest, abyśmy obserwowali inne przedkliniczne modele zaburzeń OUN, które są poddawane badaniom terapii genowej. Wcześniej badanie przedkliniczne na myszach zajmowało się terapią genową mukopolisacharydoz typu IIIA (MPS-IIIA)przez pakowanie prawidłowej wersji genu sulfamidazy wewnątrz wektora wirusowego (Sorrentino et al., 2013). W badaniu wykorzystano rosnące zrozumienie bariery krew-mózg (BBB), która aktywnie reguluje transport dużych cząsteczek z krwi do OUN w procesie zwanym transcytozą (Pardridge, 2005b; Sorrentino and Fraldi, 2016).
Transcytoza obejmuje endocytozę ligandów po stronie luminalnej, za pośrednictwem receptorów specyficznych dla ligandów (np. receptor insuliny, receptor transferyny i receptor lipoprotein o niskiej gęstości itp.,) wzbogacony na śródbłonku kapilarnym (Pardridge, 2005a). Następnie następuje przemieszczanie ładunku endocytozowanego przez cytoplazmę śródbłonka i wreszcie egzocytoza po stronie abluminalnej (mózgu), co skutecznie doprowadza ładunek przez BBB (Pardridge, 2002). W badaniu przedklinicznym MPS-IIIA wytworzono chimeryczne białko sulfamidazy, które zawierało domenę wiążącą BBB z apolipoproteiną B, aby ułatwić wychwyt przez śródbłonek, a także peptyd sygnałowy z sulfatazy iduronianowej, aby wspomóc skuteczną egzocytozę w kierunku abluminalnej strony BBB(Sorrentino et al., 2013). Ładunek wektora wirusowego kodującego chimeryczną sulfamidazę załadowano następnie do serotypu 2/8 wirusa związanego z adenami (AAV) skierowanego na wątrobę (Sorrentino and Fraldi, 2016). Tak więc wątroba służyła jako wewnętrzna fabryka, która zapewniała stałą dostawę chimeryczno-sulfamidazy, co spowodowało wzrost aktywności sulfamidazy mózgu o 10-15% nawet 7 miesięcy po terapii genowej wątroby. Ten wzrost aktywności sulfamidazy mózgu doprowadził do wymiernej poprawy patologii mózgu i wyników zachowania w mysim modelu MPS-IIIA (Sorrentino et al., 2013).
jeśli podobne badanie zostanie przeprowadzone dla INAD, odpowiedź na kilka krytycznych pytań dotyczących zastępowania enzymem terapii INAD. Alternatywnie, wewnątrz naczyniowe lub wewnątrz-CSF podawanie produktów terapii genowej opartych na AAV9 może bezpośrednio kierować na CNS (Bey i wsp ., 2017; Roca et al., 2017). Jednak liczne mutacje, które pacjenci z INAD mają na swoim Genie PLAG26, stanowią wyjątkowe wyzwania dla terapii genowej. Chociaż rozmiar genu PLA2G6 2-3 kb nie powinien stanowić problemu dla jego wstawienia do wektora wirusowego, jednak komplikacje regulacyjne wynikające z korygowania PLA2G6 są trudne do przewidzenia. Powikłania regulacyjne stają się niekontrolowane, zwłaszcza jeśli terapia genowa nie może być zlokalizowane prawidłowo w tkankach docelowych. Enzymatyczna terapia zastępcza może również stanowić problem, jeśli niektóre zmutowane produkty okażą się dominujące ujemnie w stosunku do plag26 typu dzikiego.
Większość wyzwań związanych z możliwymi terapiami INAD wynika z faktu, że jest to choroba ultra-rzadka. Jednak fakt, że INAD ma znaną etiologię genetyczną, zapewnia możliwości możliwych terapii. Co więcej, każda skuteczna terapia INAD otrzyma status leku sierocego i całą ochronę, ponieważ INAD jest rzadką chorobą. Tak więc, pomimo wszystkich wyżej wymienionych wyzwań, status leku sierocego stanowi silną zachętę dla badaczy rzadkich chorób i przemysłu biotechnologicznego, nie wspominając o tym, kiedy przyczyna genetyczna jest już znana.
Edycja genu/bazy
według stanu na 2017 r. w ludzkim Genie PLA2G6 zaobserwowano co najmniej 277 mutacji missense (Lek et al., 2016). Tylko niewielka część mutacji PLA2G6 obejmuje przesunięcia ramek, indele, mutacje nonsensowne i mutacje w miejscach splicingu (Morgan et al., 2006). Zatem korygowanie genu w populacji komórek docelowych lub korygowanie zmutowanych zasad DNA jest atrakcyjną drogą terapeutyczną. Z biegiem lat opracowano kilka narzędzi do edycji genów, a edycja genomu oparta na CRISPR/Cas9 jest okrzyknięta przełomową technologią, a badanie kliniczne planowane jest w 2018 roku. Technologie te wykorzystują białko wiążące DNA, które może również rozszczepić nici w określony sposób, aby zrobić miejsce na wstawienie nowej sekwencji DNA lub korektę szkodliwej bazy DNA(LaFountaine et al., 2015). Technologia CRISPR / Cas9 przyniosła już zdumiewające wyniki terapeutyczne w kilku przedklinicznych modelach chorób (np. dystrofia mięśniowa Duchene, choroby metaboliczne wątroby itp.,) (Dai et al., 2016). Dodatkowo, rosnące zrozumienie ludzkiej zmienności stwarza nowsze wyzwania terapii genowej i jazdy innowacji w kierunku prawdziwej personalizacji edycji genów (Lessard et al., 2017; Scott i Zhang, 2017). Nowe technologie, takie jak vslendr, wirus AAV i technologia CRISPR/Cas9 w celu zastąpienia wadliwych genów w neuronach i innych komórkach układu nerwowego, pchają technologie edycji genów do nowych granic (Nishiyama et al., 2017).
wniosek
dziecięca dystrofia Neuroaksonalna jest ciężką chorobą neurodegeneracyjną z pewną zachorowalnością i śmiertelnością. Ta rzadka choroba oferuje ekscytującą okazję do rewalidacji dostępnych trybów terapii nowej generacji i generowania nowych. Oczekuje się, że pojawiająca się zgodność z klinicznymi kryteriami diagnostycznymi dla INAD zapewni impuls w kierunku ulepszonej diagnostyki molekularnej. Oczekuje się, że postęp ten doprowadzi nas do opracowania niedrogich terapii, które zapewniłyby wymierną poprawę jakości życia pacjentów z INAD i opóźniłyby lub złagodziły postęp choroby. Podkreśliliśmy kilka sukcesów w chorobie Battena i mukopolisacharydozach, które dostarczają nam inspiracji do zadawania właściwych pytań, aby terapia INAD stała się rzeczywistością. Ponadto rosnące wirusowe i nie-wirusowe podejścia do edycji genów CRISPR/Cas9 powinny również otworzyć nowsze drogi terapeutyczne dla INAD.
wkład autora
PB i FA przygotowały wstępny projekt. SR i AC redagowały i dodały Kolejne sekcje do rękopisu. DF, AP i LP redagowały rękopis.
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. A. (1995). Hamowanie niezależnej od wapnia fosfolipazy A2 zapobiega wbudowywaniu kwasu arachidonowego i przebudowie fosfolipidów w makrofagach P388D1. Proc. Natl. Acad. Sci. U. S. A. 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Skuteczne ukierunkowanie na OUN u dorosłych myszy przez dokanałową infuzję jednoniciowego aav9-GFP do terapii genowej zaburzeń neurologicznych. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 do terapii genowej in vivo: obietnica i przeszkody. Mol. Ther. Kwasy nukleinowe 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Engel, L. A., Jing, Z., O ’ Brien, D. E., Sun, M., and Kotzbauer, P. T. (2010). Katalityczna funkcja PLA2G6 jest zaburzona przez mutacje związane z dziecięcą dystrofią neuroaksonalną, ale nie z dystonią-parkinsonizmem. PLoS One 5: e12897. doi: 10.1371 / dziennik.pone.0012897
PubMed Streszczenie / CrossRef Pełny tekst
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (1993). in PLA2G6-Associated Neurodegeneration, eds M. P. Adam, H. H. Ardinger, and R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Grzegorz, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Udział ośrodkowego układu nerwowego w chorobie Fabry’ ego: badania kliniczne i obrazowe przed i po 12 miesiącach enzymatycznej terapii zastępczej. J. Dziedzic. Metab. Dis. 27, 229–240. doi:10.1023 / B: BOLI.0000028794.04349.91
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Kapoor, S., Shah,M. H., Singh, N., raczej, M. I., Bhat, V., Gopinath, S., et al. (2016). Analiza genetyczna PLA2G6 w 22 indyjskich rodzinach z dziecięcą dystrofią neuroaksonalną, atypową późną dystrofią neuroaksonalną i zespołem dystonii parkinsonizmu. PLoS One 11: e0155605. doi: 10.1371 / dziennik.pone.0155605
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
LaFountaine, J. S., Fathe, K., and Smyth, H. D. (2015). Dostawa i zastosowania terapeutyczne technologii edycji genów ZFNs. TALENs i CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j.ijpharm.2015.08.029
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analiza genetycznej zmienności kodującej białko u 60 706 ludzi. Nature 536, 285-291. doi: 10.1038 / nature19057
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor,P. T., MacArthur, D. G., et al. (2017). Ludzka zmienność genetyczna zmienia specyficzność CRISPR-Cas9 on-i off-targeting w terapeutycznie powiązanych loci. Proc. Natl. Acad. Sci. U. S. A. 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Levi, S., and Finazzi, D. (2014). Neurodegeneracja z akumulacją żelaza w mózgu: aktualizacja mechanizmów chorobotwórczych. Przód. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Pełny tekst / Google Scholar
Liou, J. Y., Aleksic, N., Chen,S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Mitochondrialna lokalizacja cyklooksygenazy – 2 i niezależnej od wapnia fosfolipazy A2 w ludzkich komórkach nowotworowych: implikacja w oporności na apoptozę. Exp. Cela Res. 306, 75-84. doi: 10.1016 / j.yexcr.2005.01.011
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Macauley, S. L., And Sands, M. S. (2009). Obiecująca enzymatyczna terapia zastępcza ukierunkowana na OUN w chorobach przechowywania lizosomalnego. Exp. Neurol. 218, 5–8. doi: 10.1016 / j. expneurol.2009.03.040
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Mehnaaz, L. (2016). Neurodegeneracja z akumulacją żelaza w mózgu (nbia) dawniej choroba Hallervordena-Spatza. J. Assoc. Phys. Indie 64:132.
Google Scholar
2010-10-10 11: 40: 40 (2006). PLA2G6, kodujący fosfolipazę A2, jest mutowany w zaburzeniach neurodegeneracyjnych z wysoką zawartością żelaza w mózgu. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed Abstract / CrossRef Full Text / Google Scholar
Murphy, M. P. (2009). Jak mitochondria wytwarzają reaktywne formy tlenu. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstract / CrossRef Full Text / Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C., and Faria, A. (2016). Neurodegeneracja z akumulacją żelaza w mózgu: opis przypadku. Dement. Neuropsychol. 10, 160–164. doi: 10.1590 | S1980-5764-2016dn1002014
PubMed Streszczenie | CrossRef Pełny tekst / Google Scholar
Nishiyama, J., Mikuni, T., and Yasuda, R. (2017). Mediowana przez wirusa edycja genomu poprzez homologiczną naprawę w komórkach mitotycznych i postmitotycznych w mózgu ssaków. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j.neuron.2017.10.004
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., and Farooqui, A. A. (2005). Dystrybucja niezależnej od wapnia fosfolipazy A2 (iPLA2) w mózgu małp. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Pardridge, W. M. (2002). Leki i geny kierowane do mózgu molekularnymi końmi trojańskimi. Nat. Ks. Lek Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed Abstract / CrossRef Full Text / Google Scholar
Pardridge, W. M. (2005a). Biologia molekularna bariery krew-mózg. Mol. Biotechnol. 30, 57–70. pobrań: 101385 / MB:30:1:057
CrossRef Pełny tekst / Google Scholar
Pardridge, W. M. (2005b). Bariera krew-mózg: wąskie gardło w rozwoju leków w mózgu. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Fosfolipazy niezależne od wapnia A2 i ich rola w procesach biologicznych i chorobach. J. Lipiński Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Korekcja choroby za pomocą terapii genowej AAV w nowym mysim modelu mukopolisacharydozy typu IIID. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg / ddx058
PubMed Abstract / CrossRef Full Text / Google Scholar
Scott, D. A., and Zhang, F. (2017). Implikacje ludzkiej zmienności genetycznej w edycji genomu terapeutycznego opartego na CRISPR. Nat. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K., and Ma, Z. A. (2006). Niezależna od wapnia fosfolipaza A2 lokalizuje się w mitochondriach i chroni je podczas indukcji apoptotycznej przez staurosporinę. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
(2012). Terapia suplementacyjna genów dla recesywnych postaci dziedzicznych dystrofii siatkówki. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Sorrentino N. C., D ’ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Silnie wydzielana sulfamidaza zaprojektowana do przekraczania bariery krew-mózg koryguje zmiany w mózgu myszy z mukopolisacharydozami typu IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Sorrentino, N. C., and Fraldi, A. (2016). Celowanie mózgu w MPS-IIIA. Pediatr. Endokrynol. Rev. 13(Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., and Jones, S. S. (1997). Nowa cytozolowa, niezależna od wapnia fosfolipaza A2 zawiera osiem motywów ankyrinowych. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
U. S. Food and Drug Administration (2017). FDA zatwierdza pierwsze leczenie formy choroby Battena. Dostępne w: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., and Gottlieb, R. A. (2002). Hamowanie mitochondrialnej fosfolipazy A2 niezależnej od wapnia (iPLA2) łagodzi utratę mitochondrialnych fosfolipidów i jest kardioprotekcyjne. Biochem. J. 362(Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstract / CrossRef Full Text / Google Scholar
Yu-Wai-Man, P. (2016). Manipulacja genetyczna w dziedzicznych chorobach neurodegeneracyjnych: mit czy rzeczywistość? Br. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et al. (2010). Ochrona komórek beta trzustki przez grupę poprzez naprawę peroksydacji błony mitochondrialnej za pośrednictwem fosfolipazy A (2). Endocrinology 151, 3038-3048. doi: 10.1210 / en.2010-0016
PubMed Abstract / CrossRef Full Text / Google Scholar