introduktion
Infantil neuroaksonal dystrofi (INAD) er en autosomal recessiv sjælden neurodegenerativ sygdom med ukendt frekvens. Symptomdebut forekommer generelt mellem 6 måneder og 3 år. Før den tid udvikler spædbørn normalt. Det første symptom på INAD kan være at bremse opnåelsen af normale udviklingsmilepæle eller regression i udviklingsmilepæle (Ramanadham et al., 2015). Trunkhypotoni, strabismus og nystagmus er tidlige symptomer på sygdommen (Gregory et al., 2017). Progressionen af sygdommen er hurtig, og efterhånden som den skrider frem, går flere erhvervede færdigheder tabt. Muskler bliver hurtigt hypotoniske og bliver senere spastiske (Levi og Finasi, 2014). Til sidst går al frivillig muskelkontrol tabt. Muskelsvaghed kan også føre til vanskeligheder med fodring og vejrtrækning. Ud over nystagmus oplever nogle børn synstab. Kognitive funktioner går gradvist tabt, og demens udvikler sig. Levetiden er generelt 5 til 10 år (Gregory et al., 1993; Jardim et al., 2004; Macauley And Sands, 2009).
molekylær patologi
Infantil Neuroaksonal dystrofi tilhører en familie af neurodegenerative lidelser, der omfatter atypisk sen-onset neuroaksonal dystrofi (ANAD) og dystoni Parkinsonisme kompleks (DPC). De fleste tilfælde af INAD er forbundet med homosygøse eller sammensatte heterosygøse mutationer i PLA2G6-genet, der påvirker den katalytiske aktivitet af dets proteinprodukt (Engel et al., 2010). PLA2G6-genet koder for en gruppe via calciumuafhængig phospholipase A2-protein (PLA2G6 eller ipla2 Lira, 85/88 kDa) med en lipase og syv ankyrin-gentagelsesholdige domæner (Tang et al., 1997). PLA2G6 hydrolyserer SN-2 acylkæden af phospholipider, der genererer frie fedtsyrer og lysophospholipider.
phospholipider i mitokondriernes indre membran er rige på umættede fedtsyrer I SN-2-positionen, især cardiolipin (Selesnev et al., 2006). Disse umættede fedtsyrer er særligt sårbare over for de rigelige reaktive iltarter produceret af mitokondrier (Murphy, 2009), hvilket resulterer i peroksidiserede phospholipider i mitokondriernes indre membran. PLA2G6 lokaliseres til mitokondrier (2002; Liou et al., 2005) i overensstemmelse med en øget efterspørgsel efter hydrolyse af de peroksidiserede fedtsyrer i sn-2-positionen af phospholipider, der fører til ombyggede phospholipider (Balsinde et al., 1995; Jens et al., 2010). Når PLA2G6 er defekt, er mitokondriernes indre membranintegritet beskadiget. PLA2G6 lokaliseres også til akson (Ong et al., 2005; Selesnev et al., 2006), der også indikerer en øget lokal efterspørgsel efter phospholipid-ombygning der. Manifestationen af en sådan ophobning i hjernen er unik for centrale hjerneområder, såsom de basale ganglier, hvilket resulterede i forskellige navne for den samme underliggende molekylære patogenese, der involverede PLAG26 (Mehnaas, 2016; Nassif et al., 2016).
Ultrastrukturanalyse af neuroner i PLA2G6 knockout mus er i overensstemmelse med denne molekylære patologi. Mitokondrier med forgrening og rørformet cristae, mitokondrier med degenereret cristae, aksoner med cytoskelet kollaps og delvis membrantab ved aksonterminaler er blevet observeret (Beck et al., 2011). På et mikroskopisk niveau fremstår disse træk som aksonale hævelser og sfæroide legemer i præsynaptiske terminaler (Figur 1) i det centrale eller perifere nervesystem.
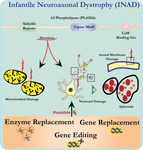
Figur 1. Infantil neuroaksonal dystrofi (INAD) er en neurodegenerativ lidelse relateret til mutationer i PLA2G6-genet. Forskellige mutationer af PLA2G6 fører til dysfunktionel A2-phospholipase, der fører til mitokondrie-og aksonale membranfejl. Disse defekter forårsager neuronal skade visualiseret som aksonale hævelser og ophobning af præsynaptiske sfæroider. For at gendanne funktioner, genudskiftning eller redigering for at korrigere den defekte PLA2G6 foreslås terapeutiske strategier.
molekylær diagnostik og Patientindflydelse med sjælden sygdom
bortset fra specifikke kliniske, elektrofysiologiske og billeddannende funktioner, før tilgængeligheden af næste generations sekventering, hudbiopsier, der viser aksonale hævelser og sfæroide kroppe i præsynaptiske terminaler i det centrale eller perifere nervesystem var de diagnostiske kriterier for bekræftelse af INAD (Gregory et al., 2017; Iodice et al., 2017). Ofte var der behov for flere biopsier for at bekræfte diagnosen. Familier ventede generelt mange år på en diagnose. Med de faldende omkostninger ved gen-og genomsekventering, tilgængelighed af målrettet genpaneltest med diagnostiske laboratorier for udiagnosticerede neurologiske sygdomme, og den stigende opmærksomhed hos læger om tilgængeligheden af genetisk diagnostik, familier modtager diagnose hurtigere; nogle gange inden for et år efter det første symptom vises. Børnene i disse familier er stadig unge, og familierne er motiverede til at samarbejde med forskere for at finde en behandling for deres børns sygdom. For at finansiere forskningsprocessen dannede en gruppe forældre til INAD-patienter INADcure Foundation. Fonden har skaffet betydelige midler til forskning og samarbejder med Rare Genomics Institute for at vejlede dem i tildelingen af forskningsstipendier. Interessant nok fandt en genetisk analyse fra 2016 af 22 indiske familier med INAD, ANAD og DPC, at 10/22 familier (45,45%) manglede mutationer i PLA2G6-genkodningsregionen (Kapoor et al., 2016). Manglende identifikation af skadelige mutationer i det kodende område af PLA2G6 fremhæver, at fremtidig molekylær diagnostisk indsats ville kræve hel gensekventering for at identificere mutationer i de introniske og regulatoriske regioner i PLA2G6-genet hos INAD-berørte patienter. Desuden antyder INAD-tilfælde, hvor PLA2G6-associerede mutationer ikke observeres, at årsagen til sygdommen kan skyldes mutation i andre gener end PLA2G, som skal undersøges.
mulige terapier
de fleste af de terapier, der overvejes for en sjælden sygdom som INAD, der involverer et defekt f.eks. Når en recessiv genetisk defekt er forårsaget af en recessiv genetisk defekt, antages det, at erstatning eller tilskud kan rette op på problemet (Smith et al., 2012, 2016). Imidlertid, eksperimenter for at bevise, at terapier, der leverer det korrekte gen eller f.eks. vil redde Inad-fænotypen, skal endnu ikke udføres og testes.
ERT (ERT)
da hjernen er det primære organ, der er påvirket af INAD, vil Inad sandsynligvis kræve infusion af Inad i hjernen. I 2017 modtog Biomarin FDA-godkendelse af tripeptidylpeptidase 1 (cerliponase alfa) som en behandling for den underliggende årsag til Batten-sygdom, manglen på tripeptidylpeptidase (TPP1). Tripeptidylpeptidase 1 (cerliponase alfa; 59 kDa) er den første ERT, der administreres direkte i hjernens cerebrospinalvæske (CSF). Andre ERT-lægemidler, der kan administreres i CSF, er i kliniske forsøg (Jardim et al., 2004; Macauley And Sands, 2009).
effekten af tripeptidylpeptidase 1 (cerliponase alfa) på gangsevnen hos patienter med Batten-sygdom viser, at erstatningsterapi for sygdomme, der påvirker hjernen, er teoretisk mulig. Inad vil være mere udfordrende end lysosomal målretning, som allerede havde været vellykket af IV ERT i lang tid som i tilfælde af Gauchers sygdom. Derfor er der stadig mange biologiske problemer, der er specifikke for INAD, der skal løses:
• hvilken mekanisme kan bruges til muligvis at lokalisere PLA2G6 til mitokondrier?
• er der et særligt behov for at adressere målretningen af det perifere nervesystem?
• kan en delvis PLA2G6 redde dens mangel, eller kræves et helt protein?
• hvad er et passende opløsningsmiddel til levering af PLA2G6-genproduktet i CSF?
• hvis PLA2G6 ikke er 100% ren, Hvad er dens forurenende stoffer, og er de sikre?
• vil der være generaliserede intravaskulære komplikationer på grund af pla2g6-infusion i CNS? Har infusionsdosis nogen relevans for sådanne komplikationer?
et yderligere problem er, at erstatningsterapi kræver den første placering af et intracerebroventrikulært kateter og hyppige infusioner. Placeringen af kateteret kræver anæstesi, og infusionerne kan kræve anæstesi afhængigt af patienternes samarbejde. At tackle bekymringerne hos pædiatriske neurologer og forældre til børn med INAD med hensyn til brug af anæstesi på INAD-patienter med passende information vil kræve yderligere indsats.
genterapi/Generstatning
det humane PLA2G-gen er omkring 70 kb med 17 eksoner og 2 alternative eksoner. Det længste proteinkodende transkript er dog kun 3,3 kb, og proteinkodningssekvensen er lidt over 2,4 kb, som let kan pakkes ind i en viral vektorlast. Vi vil først undersøge en anden lysosomal opbevaringsforstyrrelse i CNS, der har prækliniske data, der derefter skal sammenlignes med INAD. Manglen på prækliniske data for ERT i INAD er et betydeligt vidensgab på området, hvorfor det er afgørende, at vi observerer andre prækliniske modeller af CNS-lidelser, der gennemgår genterapiundersøgelser. Tidligere behandlede en præklinisk undersøgelse hos mus genterapi for lysosomal opbevaringsforstyrrelse mucopolysaccharidoses type IIIA (MPS-IIIa) ved at pakke den korrekte version af sulfamidase-genet inde i en viral vektor (Sorrentino et al., 2013). Undersøgelsen udnyttede den voksende forståelse af blodhjernebarrieren (BBB), som aktivt regulerer transporten af store molekyler fra blod til CNS ved en proces kaldet transcytose (Pardridge, 2005B; Sorrentino og Fraldi, 2016).
Transcytose involverer endocytose af ligander på luminalsiden, medieret af ligandspecifikke receptorer (f.eks. insulinreceptor, transferrinreceptor og lipoproteinreceptor med lav densitet osv.,) beriget på kapillærendotelet (Pardridge, 2005A). Dette efterfølges af bevægelse af endocytoseret last gennem endotel-cytoplasmaet og endelig eksocytose ved abluminal (hjerne) side og leverer således effektivt lasten over BBB (Pardridge, 2002). MPS-IIIa præklinisk undersøgelse fremstillede et kimært sulfamidaseprotein, der indeholdt et BBB-bindende domæne fra apolipoprotein B for at lette optagelsen af endotelet og også et signalpeptid fra iduronat-2-sulfatase for at hjælpe en effektiv eksocytose mod den abluminale side af BBB (Sorrentino et al., 2013). En viral vektorlast, der koder for en kimær sulfamidase, blev derefter indlæst på en adenoassocieret virus (AAV) serotype 2/8 rettet mod leveren (Sorrentino og Fraldi, 2016). Leveren tjente således som en intern fabrik, der tilvejebragte en konstant forsyning af kimærsulfamidase, hvilket resulterede i en 10-15% stigning i hjernesulfamidaseaktivitet selv 7 måneder efter levergenterapi. Denne stigning i hjernesulfamidaseaktivitetsniveauer førte til kvantificerbar forbedring i hjernepatologi og adfærdsresultater i musemodellen af MPS-IIIA (Sorrentino et al., 2013).
hvis en lignende undersøgelse udføres for INAD, ville den besvare flere kritiske spørgsmål om erstatning for INAD-terapi. Alternativt kan intra-vaskulær eller intra-CSF-administration af aav9-baserede genterapiprodukter direkte målrette CNS (Bey et al., 2017; Roca et al., 2017). Imidlertid giver de mange mutationer, som INAD-patienter har på deres PLAG26-gen, unikke udfordringer for genterapi. Selvom størrelsen af PLA2G6-genet på 2-3 kb ikke bør udgøre et problem for dets indsættelse i en viral vektor, er regulatoriske komplikationer fra korrigering af PLA2G6 imidlertid vanskelige at forudsige. Regulatoriske komplikationer bliver ukontrollerbare, især hvis genterapien ikke kan lokaliseres korrekt inden for målvævene. Erstatningsterapi kan også være et problem, hvis nogle mutante produkter viser sig at være dominerende negative for vildtype PLAG26.
de fleste af udfordringerne ved mulige terapier for INAD kommer fra det faktum, at det er en ultra-sjælden sygdom. Det faktum, at INAD har en kendt genetisk etiologi, giver imidlertid muligheder for mulige terapier. Desuden vil enhver vellykket terapi for INAD modtage forældreløs lægemiddelstatus og al den beskyttelse, den får, fordi INAD er en sjælden sygdom. På trods af alle de førnævnte udfordringer giver status for forældreløse lægemidler et stærkt incitament for forskere i sjældne sygdomme og bioteknologiindustrien, for ikke at nævne, når den genetiske årsag allerede er kendt.
Gen/Baseredigering
fra 2017 er der observeret mindst 277 missense-mutationer i det humane PLA2G6-gen (Lek et al., 2016). Kun en lille del af PLA2G6-mutationer inkluderer rammeskift, indels, nonsensmutationer og mutationer i splejsningsstederne (Morgan et al., 2006). Korrigering af genet i målcellepopulationen eller korrigering af de muterede DNA-baser er således en attraktiv terapeutisk vej. I årenes løb er der udviklet flere værktøjer til genredigering, og CRISPR/Cas9-baseret genomredigering hyldes som en banebrydende teknologi med et klinisk forsøg planlagt i 2018. Disse teknologier anvender et DNA-bindende protein, der også kan spalte strengen på en specificeret måde for at give plads til indsættelse af en ny DNA-sekvens eller korrektion af den skadelige DNA-base (LaFountaine et al., 2015). CRISPR / Cas9-teknologi har allerede givet forbløffende terapeutiske resultater i flere prækliniske sygdomsmodeller (f.eks.,) (Dai et al., 2016). Derudover udgør en voksende forståelse af menneskelig variation nyere udfordringer for genterapi og driver innovation mod en ægte personalisering af genredigering (Lessard et al., 2017; Scott og Jang, 2017). Nye teknologier såsom vSLENDR, en AAV-virus og CRISPR/Cas9-medieret teknologi til erstatning af defekte gener i neuroner og andre nervesystemceller skubber genredigeringsteknologier til nye grænser (Nishiyama et al., 2017).
konklusion
Infantil Neuroaksonal dystrofi er en alvorlig neurodegenerativ sygdom med en vis sygelighed og dødelighed. Denne sjældne sygdom giver en spændende mulighed for at revalidere tilgængelige former for næste generations Terapi og generere nyere. Emerging kongruens på de kliniske diagnostiske kriterier for INAD forventes at give en drivkraft mod forbedret molekylær diagnostik. Disse fremskridt forventes at føre os til at udvikle overkommelige terapier, der ville give kvantificerbar forbedring af livskvaliteten for INAD-patienter og forsinke eller forbedre sygdomsprogressionen. Vi har fremhævet nogle succeshistorier i Batten ‘ s sygdom og mucopolysaccharidoses, der giver os inspiration til at stille de rigtige spørgsmål for at gøre INAD-terapi til virkelighed. Derudover bør voksende virale og ikke-virale tilgange til CRISPR/Cas9-baseret genredigering også åbne nyere terapeutiske veje for INAD.
Forfatterbidrag
PB og FA udarbejdede foreløbigt udkast. SR og AC redigerede og tilføjede flere sektioner til manuskriptet. DF, AP og LP redigerede manuskriptet.
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K. og Dennis, E. A. (1995). Inhibering af calciumuafhængig phospholipase A2 forhindrer inkorporering af arachidonsyre og ombygning af phospholipid i p388d1-makrofager. Proc. Natl. Acad. Sci. USA 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Effektiv CNS-målretning hos voksne mus ved intratekal infusion af enkeltstrenget AAV9-GFP til genterapi af neurologiske lidelser. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
dai, J.-J., Ju, L.-Y., Yan, Y.-Y., Y., Vang, K.-L., Lu, K.-J., et al. (2016). CRISPR-Cas9 til in vivo genterapi: løfte og forhindringer. Mol. Ther. Nukleinsyrer 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Engel, L. A., Jing, O ‘ Brien, D. E. P. T., P. T. (2010). Katalytisk funktion af PLA2G6 er svækket af mutationer forbundet med infantil neuroaksonal dystrofi, men ikke dystoni-parkinsonisme. PLoS en 5: e12897. doi: 10.1371 / tidsskrift.pone.0012897
PubMed abstrakt / CrossRef Fuld tekst
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P. og Hayflick, S. J. (1993). i PLA2G6-associeret Neurodegeneration, eds M. P. Adam, H. H. Ardingerog R. A. Pagon, Seattle, var: Generevisninger.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). CNS-involvering i Fabrys sygdom: kliniske undersøgelser og billeddannelsesundersøgelser før og efter 12 måneders erstatningsterapi. J. Arve. Metab. Dis. 27, 229–240. doi: 10.1023 / B: BOLI.0000028794.04349.91
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., snarere, M. I., Bhat, V., Gopinath, S., Et Al. (2016). Genetisk analyse af PLA2G6 i 22 indiske familier med infantil neuroaksonal dystrofi, atypisk sen-onset neuroaksonal dystrofi og dystoni parkinsonisme kompleks. PLoS en 11: e0155605. doi: 10.1371 / tidsskrift.pone.0155605
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
LaFountaine, J. S., Fathe, K. og Smyth, H. D. (2015). Levering og terapeutiske anvendelser af genredigeringsteknologier. TALENs og CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., Et Al. (2016). Analyse af proteinkodende genetisk variation hos 60.706 mennesker. Natur 536, 285-291. doi: 10.1038 / nature19057
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Human genetisk variation ændrer CRISPR – Cas9 on-og off-targeting specificitet på terapeutisk implicerede loci. Proc. Natl. Acad. Sci. U. S. A. 14, E11257–E11266. doi: 10.1073 / pnas.1714640114
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Levi, S., D. (2014). Neurodegeneration med hjernejernakkumulering: opdatering om patogene mekanismer. Front. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Fuld tekst / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., K. K., Et Al. (2005). Mitokondriel lokalisering af cyclooksygenase – 2 og calciumuafhængig phospholipase A2 i humane kræftceller: implikation i apoptoseresistens. Eksp. Celle Res. 306, 75-84. doi: 10.1016 / j.2005.01.011
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Macauley, S. L. og Sands, M. S. (2009). Lovende CNS-rettet erstatningsterapi til lysosomale opbevaringssygdomme. Eksp. Neurol. 218, 5–8. doi: 10.1016 / j.2009.03.040
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Mehna, L. (2016). Neurodegeneration med hjernejernakkumulering (NBIA) tidligere Hallervorden – Spatts sygdom. J. Assoc. Phys. Indien 64: 132.
Google Scholar
det er en af de bedste måder at gøre dette på. (2006). PLA2G6, der koder for en phospholipase A2, muteres i neurodegenerative lidelser med højt hjernejern. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Murphy, M. P. (2009). Hvordan mitokondrier producerer reaktive iltarter. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Nassif, D., Pereira, J. S., spids, M., Capit Larso, C. og Faria, A. (2016). Neurodegeneration med hjernejernakkumulering: en sagsrapport. Dement. Neuropsykol. 10, 160–164. doi: 10.1590 / S1980-5764-2016dn1002014
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
Nishiyama, J., Mikuni, T. og Yasuda, R. (2017). Virus-medieret genomredigering via homologi-rettet reparation i mitotiske og postmitotiske celler i pattedyrs hjerne. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j. neuron.2017.10.004
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
i 2005 blev der lavet en ny bog, der blev udgivet af A. A. (engelsk). Fordeling af calciumuafhængig phospholipase A2 (iPLA2) i abehjernen. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Pardridge, M. M. (2002). Narkotika og gen rettet mod hjernen med molekylære trojanske heste. Nat. Rev. Drug Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Pardridge, M. M. (2005A). Molekylærbiologi af blod-hjernebarrieren. Mol. Biotechnol. 30, 57–70. doi: 10.1385 / MB:30: 1:057
CrossRef Fuld tekst | Google Scholar
Pardridge, H. M. (2005B). Blod-hjerne-barrieren: flaskehals i hjernens lægemiddeludvikling. Neurorks 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. V., Bone, R. N., Hancock, L. D., Lei, et al. (2015). Calciumuafhængige phospholipaser A2 og deres roller i biologiske processer og sygdomme. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Roca, C., Motas, S., Marc Kurt, S., Ribera, A., S, V., S, et al. (2017). Sygdomskorrektion ved AAV-medieret genterapi i en ny musemodel af mucopolysaccharidose type IIID. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg / ddks058
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Scott, D. A. og Jang, F. (2017). Implikationer af menneskelig genetisk variation i CRISPR-baseret terapeutisk genomredigering. Nat. Middelhavs. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Selesnev, K., Chao, C., Chang, H. Song, K. og Ma (2006). Calciumuafhængig phospholipase A2 lokaliserer i og beskytter mitokondrier under apoptotisk induktion af staurosporin. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Smith, A. J., Bainbridge, J. V. og Ali, R. R. (2012). Gentilskudsterapi til recessive former for arvelige retinale dystrofier. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Sorrentino, N. C., D ‘ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., Et Al. (2013). En stærkt udskilt sulfamidase konstrueret til at krydse blod-hjerne-barrieren korrigerer hjernelæsioner hos mus med mucopolysaccharidoser type IIIA. EMBO Mol. Middelhavs. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Sorrentino, N. C. og Fraldi, A. (2016). Hjernemålretning i MPS-IIIA. Pediatr. Endocrinol. Åb 13 (Suppl. 1), 630–638.
Google Scholar
Tang, J., Kris, R. V., Ulvemand, N., Shaffer, M., Seehra, J. og Jones, S. S. (1997). En ny cytosolisk calciumuafhængig phospholipase A2 indeholder otte ankyrinmotiver. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Food and Drug Administration (2017). FDA godkender første behandling for en Form for Batten sygdom. Tilgængelig på: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
S. D. og Gottlieb, R. A. (2002). Inhibering af mitokondrie calcium-uafhængig phospholipase A2 (iPLA2) dæmper mitokondrie phospholipid tab og er hjertebeskyttende. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Yu-vai-Man, P. (2016). Genetisk manipulation for arvelige neurodegenerative sygdomme: myte eller virkelighed? Br. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Choi, J., Shi, J., Song, K., et al. (2010). Beskyttelse af pancreasbetaceller efter gruppe via phospholipase A(2)-medieret reparation af mitokondriel membranperoksidering. Endokrinologi 151, 3038-3048. doi: 10.1210 / da.2010-0016
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar