introduktion
Infantil neuroaxonal dystrofi (INAD) är en autosomal recessiv sällsynt neurodegenerativ sjukdom med okänd frekvens. Uppkomsten av symtom uppträder vanligtvis mellan 6 månader och 3 år. Före den tiden utvecklas spädbarn normalt. Det första symptomet på INAD kan vara att sakta ner uppnåendet av normala utvecklingsmilstoler eller regression i utvecklingsmilstoler (Ramanadham et al., 2015). Stamhypotoni, strabismus och nystagmus är tidiga symtom på sjukdomen (Gregory et al., 2017). Sjukdomsprogressionen är snabb och när den fortskrider förloras mer förvärvade färdigheter. Muskler blir snart hypotoniska och blir senare spastiska (Levi och Finazzi, 2014). Så småningom förloras all frivillig muskelkontroll. Muskelsvaghet kan också leda till svårigheter att mata och andas. Förutom nystagmus upplever vissa barn synförlust. Kognitiva funktioner förloras gradvis och demens utvecklas. Livslängden är i allmänhet 5 till 10 år (Gregory et al., 1993; Jardim et al., 2004; Macauley och Sands, 2009).
Molekylär patologi
Infantil Neuroaxonal dystrofi tillhör en familj av neurodegenerativa störningar som inkluderar atypisk sen debut neuroaxonal dystrofi (ANAD) och dystoni Parkinsonism complex (DPC). De flesta fall av INAD är associerade med homozygota eller sammansatta heterozygota mutationer i PLA2G6-genen som påverkar den katalytiska aktiviteten hos dess proteinprodukt (Engel et al., 2010). PLA2G6-genen kodar för en grupp via kalciumoberoende fosfolipas A2-protein (PLA2G6 eller ipla2 msk, 85/88 kDa, 85/88 kDa) med ett lipas och sju ankyrin-upprepande domäner (Tang et al., 1997). PLA2G6 hydrolyserar SN-2-acylkedjan av fosfolipider och genererar fria fettsyror och lysofosfolipider.
fosfolipider i mitokondriernas inre membran är rika på omättade fettsyror i sn-2-positionen, särskilt kardiolipin (Seleznev et al., 2006). Dessa omättade fettsyror är särskilt sårbara för de rikliga reaktiva syrearterna som produceras av mitokondrierna (Murphy, 2009) vilket resulterar i peroxidiserade fosfolipider i mitokondriernas inre membran. PLA2G6 lokaliseras till mitokondrier (Williams och Gottlieb, 2002; Liou et al., 2005) överensstämmer med en ökad efterfrågan på hydrolys av de peroxiderade fettsyrorna i sn-2-positionen för fosfolipider som leder till ombyggda fosfolipider (Balsinde et al., 1995; Zhao et al., 2010). När PLA2G6 är defekt skadas mitokondriernas inre membranintegritet. PLA2G6 lokaliseras också till axonen (Ong et al., 2005; Seleznev et al., 2006) indikerar en ökad lokal efterfrågan på fosfolipid ombyggnad där också. Manifestationen av sådan ackumulering i hjärnan är unik för viktiga hjärnområden, såsom basala ganglier som resulterade i olika namn för samma underliggande molekylära patogenes som involverar PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
Ultrastrukturanalys av neuroner i PLA2G6 knockout-möss överensstämmer med denna molekylära patologi. Mitokondrier med förgrening och tubulär cristae, mitokondrier med degenererad cristae, axoner med cytoskeletonkollaps och partiell membranförlust vid axonterminaler har observerats (Beck et al., 2011). På mikroskopisk nivå framträder dessa egenskaper som axonala svullnader och sfäroidkroppar i presynaptiska terminaler (Figur 1) i centrala eller perifera nervsystemet.
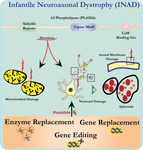
Figur 1. Infantil neuroaxonal dystrofi (INAD) är en neurodegenerativ störning relaterad till mutationer i PLA2G6-genen. Olika mutationer av PLA2G6 leder till dysfunktionellt A2-fosfolipas som leder till mitokondriella och axonala membranfel. Dessa defekter orsakar neuronal skada visualiserad som axonala svullnader och ackumulering av presynaptiska sfäroider. Enzymbyte för att återställa funktioner, genbyte eller redigering för att korrigera den defekta PLA2G6 föreslås terapeutiska strategier.
molekylär diagnostik och sällsynt sjukdom Patient Empowerment
bortsett från specifika kliniska, elektrofysiologiska och avbildningsfunktioner, före tillgängligheten av nästa generations sekvensering, hudbiopsier som visar axonala svullnader och sfäroidkroppar i presynaptiska terminaler i centrala eller perifera nervsystemet var de diagnostiska kriterierna för bekräftelse av INAD (Gregory et al., 2017; Jodice et al., 2017). Ofta krävdes flera biopsier för att bekräfta diagnosen. Familjer väntade i allmänhet många år på en diagnos. Med den minskande kostnaden för gen-och genomsekvensering, tillgänglighet av riktad genpaneltestning med diagnostiska laboratorier för odiagnostiserade neurologiska sjukdomar och den ökande medvetenheten hos läkare om tillgängligheten av genetisk diagnostik får familjer diagnosen snabbare; ibland inom ett år efter det första symptomet. Barnen i dessa familjer är fortfarande unga och familjerna är motiverade att samarbeta med forskare för att hitta en behandling för sina barns sjukdom. För att finansiera forskningsprocessen bildade en grupp föräldrar till INAD-patienter INADcure Foundation. Stiftelsen har samlat in betydande medel för forskning och samarbetar med Rare genomics Institute för att vägleda dem vid tilldelning av forskningsbidrag. Intressant nog fann en 2016 genetisk analys av 22 indiska familjer med INAD, ANAD och DPC att 10/22 familjer (45,45%) saknade mutationer i PLA2G6-genkodningsregionen (Kapoor et al., 2016). Underlåtenhet att identifiera skadliga mutationer i den kodande regionen PLA2G6 belyser att framtida molekylära diagnostiska ansträngningar skulle kräva hel gensekvensering för att identifiera mutationer i de introniska och reglerande regionerna i PLA2G6-genen hos INAD-drabbade patienter. Vidare antyder INAD-fall där PLA2G6-associerade mutationer inte observeras att orsaken till sjukdomen kan bero på mutation i andra gener än PLA2G, som måste undersökas.
möjliga terapier
de flesta terapier som övervägs för en sällsynt sjukdom som INAD som involverar ett defekt enzym är enzymersättning, genersättning eller genkorrigering. När en enzymbrist orsakas av en recessiv genetisk defekt antas det att enzymersättning eller tillskott kan korrigera problemet (Smith et al., 2012; Yu-Wai-Man, 2016). Men experiment för att bevisa att terapier som ger rätt gen eller enzym kommer att rädda Inad-fenotypen ännu inte ska utföras och testas.
Enzymersättningsterapi (ERT)
eftersom hjärnan är det primära organ som påverkas i INAD, skulle enzymersättningsterapi för INAD sannolikt kräva infusion av enzymet i hjärnan. I 2017 mottog Biomarin FDA-godkännande för tripeptidylpeptidas 1 (cerliponase alfa) som en behandling för den bakomliggande orsaken till Batten sjukdom, bristen på tripeptidylpeptidas (TPP1) ett lysosomalt enzym (US Food Drug Administration, 2017). Tripeptidylpeptidas 1 (cerliponas alfa; 59 kDa) är den första ERT som administreras direkt i cerebrospinalvätskan (CSF) i hjärnan. Andra ERT-läkemedel som kan administreras i CSF är i kliniska prövningar (Jardim et al., 2004; Macauley och Sands, 2009).
effekten av tripeptidylpeptidas 1 (cerliponase alfa) på gångförmågan hos Batten-sjukdomspatienter visar att enzymersättningsterapi för sjukdomar som påverkar hjärnan är teoretiskt möjligt. Inriktning på ersättningsenzym i mitokondrier för INAD kommer att vara mer utmanande än lysosomal inriktning som redan hade varit framgångsrik av IV ERT under lång tid som i fallet med Gauchers sjukdom. Därför finns det fortfarande många biologiska problem som är specifika för INAD som måste lösas:
• behöver terapi för INAD mekanismer som eventuellt kan transportera PLA2G6 in i cellen?
en ytterligare fråga är att enzymersättningsterapi kräver initial placering av en intracerebroventrikulär kateter och frekventa infusioner. Placeringen av katetern kräver anestesi och infusionerna kan kräva anestesi beroende på patienternas samarbete. Att ta itu med oro hos pediatriska neurologer och föräldrar till barn med INAD när det gäller att använda anestesi på INAD-patienter med lämplig information kommer att kräva ytterligare ansträngningar.
genterapi/Genersättning
den humana PLA2G-genen är cirka 70 kb med 17 exoner och 2 alternativa exoner. Det längsta proteinkodande transkriptet är dock bara 3,3 kb och proteinkodningssekvensen är drygt 2,4 kb, som lätt kan förpackas i en viral vektorlast. Vi kommer först att utforska en annan lysosomal lagringsstörning i CNS som har prekliniska data för att sedan jämföras med INAD. Bristen på prekliniska data för ERT i INAD är ett betydande kunskapsgap inom området, varför det är avgörande att vi observerar andra prekliniska modeller av CNS-störningar som genomgår genterapistudier. Tidigare behandlade en preklinisk studie på möss genterapi för den lysosomala lagringsstörningen mukopolysackaridoser typ IIIA (MPS-IIIA) genom att förpacka den korrekta versionen av sulfamidasgenen inuti en viral vektor (Sorrentino et al., 2013). Studien utnyttjade den växande förståelsen av blodhjärnbarriären (BBB) som aktivt reglerar transporten av stora molekyler från blod till CNS genom en process som kallas transcytos (Pardridge, 2005B; Sorrentino och Fraldi, 2016).
Transcytos involverar endocytos av ligander på luminalsidan, medierad av ligandspecifika receptorer (t.ex. insulinreceptor, transferrinreceptor och lågdensitetslipoproteinreceptor, etc.,) berikad på kapillärendotelet (Pardridge, 2005a). Detta följs av rörelse av endocytoserad Last genom endotelcytoplasman och slutligen exocytos vid abluminal (hjärna) sida, vilket effektivt levererar lasten över BBB (Pardridge, 2002). Mps-IIIA preklinisk studie tillverkade ett chimärt sulfamidasprotein som innehöll en BBB-bindande domän från apolipoprotein B för att underlätta upptag av endotelet och även en signalpeptid från iduronat-2-sulfatas för att underlätta en effektiv exocytos mot abluminalsidan av BBB (Sorrentino et al., 2013). En viral vektorlast som kodar för ett chimärt sulfamidas laddades sedan på en adenoassocierad virus (AAV) serotyp 2/8 riktad mot levern (Sorrentino och Fraldi, 2016). Således fungerade levern som en intern fabrik som gav en konstant tillförsel av chimär-sulfamidas, vilket resulterade i en 10-15% ökning av hjärnans sulfamidasaktivitet även 7 månader efter levergenterapi. Denna ökning av hjärnsulfamidasaktivitetsnivåer ledde till kvantifierbar förbättring av hjärnpatologi och beteenderesultat i musmodellen för MPS-IIIA (Sorrentino et al., 2013).
om en liknande studie utförs för INAD, skulle den svara på flera kritiska frågor om enzymersättning för INAD-terapi. Alternativt kan intra-vaskulär eller intra-CSF-administrering av aav9-baserade genterapiprodukter direkt rikta sig mot CNS (Bey et al., 2017; Roca et al., 2017). De multipla mutationer som INAD-patienter har på sin PLAG26-gen ger emellertid unika utmaningar för genterapi. Även om storleken på PLA2G6-genen på 2-3 kb inte borde utgöra ett problem för dess införande i en viral vektor, är de regulatoriska komplikationerna från att korrigera PLA2G6 svåra att förutsäga. Regulatoriska komplikationer blir okontrollerbara, särskilt om genterapin inte kan lokaliseras ordentligt i målvävnaderna. Enzymersättningsterapi kan också vara ett problem om vissa mutanta produkter visar sig vara dominerande negativa för vildtypen PLAG26.
de flesta av utmaningarna med möjliga terapier för INAD kommer från det faktum att det är en extremt sällsynt sjukdom. Det faktum att INAD har en känd genetisk etiologi ger emellertid vägar för möjliga terapier. Dessutom kommer varje framgångsrik behandling för INAD att få särläkemedelsstatus och allt skydd Det får eftersom INAD är en sällsynt sjukdom. Trots alla ovannämnda utmaningar ger särläkemedelsstatusen ett starkt incitament för sällsynta sjukdomsforskare och bioteknikindustrin, för att inte tala om när den genetiska orsaken redan är känd.
Gen/Basredigering
från och med 2017 har minst 277 missense-mutationer observerats i den humana PLA2G6-genen (Lek et al., 2016). Endast en liten del av PLA2G6-mutationer inkluderar ramförskjutningar, indels, nonsensmutationer och mutationer i skarvplatserna (Morgan et al., 2006). Således är korrigering av genen i målcellpopulationen eller korrigering av de muterade DNA-baserna en attraktiv terapeutisk aveny. Under åren har flera verktyg utvecklats för genredigering, och CRISPR/Cas9-baserad genomredigering hyllas som en banbrytande teknik med en klinisk prövning planerad 2018. Dessa tekniker använder ett DNA-bindande protein som också kan klyva strängen på ett specificerat sätt för att göra plats för införandet av en ny DNA-sekvens eller korrigering av den skadliga DNA-basen (LaFountaine et al., 2015). CRISPR / Cas9-tekniken har redan gett häpnadsväckande terapeutiska resultat i flera prekliniska sjukdomsmodeller (t.ex. Duchene muskeldystrofi, levermetaboliska sjukdomar etc.,) (Dai et al., 2016). Dessutom utgör en växande förståelse för mänsklig variation nyare utmaningar för genterapi och driver innovation mot en sann personalisering av genredigering (Lessard et al., 2017; Scott och Zhang, 2017). Ny teknik som vSLENDR, ett AAV-virus och CRISPR/Cas9-medierad teknik för att ersätta defekta gener i neuroner och andra nervsystemceller Driver genredigeringsteknik till nya gränser (Nishiyama et al., 2017).
slutsats
Infantil Neuroaxonal dystrofi är en allvarlig neurodegenerativ sjukdom med viss sjuklighet och dödlighet. Denna sällsynta sjukdom erbjuder en spännande möjlighet att förnya tillgängliga lägen för nästa generations Terapi och generera nyare. Framväxande kongruens på de kliniska diagnostiska kriterierna för INAD förväntas ge en drivkraft mot förbättrad molekylär diagnostik. Dessa framsteg förväntas leda oss till att utveckla prisvärda terapier som skulle ge kvantifierbar förbättring av livskvaliteten hos INAD-patienter och fördröja eller förbättra sjukdomsprogressionen. Vi har belyst några framgångshistorier i Bats sjukdom och mukopolysackaridoserna som ger oss inspiration att ställa rätt frågor för att göra INAD-terapi till verklighet. Dessutom bör växande virala och icke-virala tillvägagångssätt för CRISPR/Cas9-baserad genredigering också öppna nyare terapeutiska vägar för INAD.
författare bidrag
PB och FA beredd preliminära utkast. SR och AC redigerade och lade till fler avsnitt i manuskriptet. DF, AP och LP redigerade manuskriptet.
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
Balsinde, J., Bianco, I. D., Ackermann, Ej, Conde-Frieboes, K. och Dennis, E. A. (1995). Hämning av kalciumoberoende fosfolipas A2 förhindrar arakidonsyrainkorporering och fosfolipidremodellering i p388d1-makrofager. Proc. Natl. Acad. Sci. USA 92, 8527-8531. doi: 10.1073/pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Effektiv CNS-inriktning hos vuxna möss genom intratekal infusion av enkelsträngad AAV9-GFP för genterapi av neurologiska störningar. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstrakt / CrossRef fulltext / Google Scholar
dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 för in vivo genterapi: löfte och hinder. Mol. Där. Nukleinsyror 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Engel, L. A., Jing, Z., O ’ Brien, D. E., Sun, M. och Kotzbauer, P. T. (2010). Katalytisk funktion av PLA2G6 försämras av mutationer associerade med infantil neuroaxonal dystrofi men inte dystoni-parkinsonism. PLoS en 5: e12897. doi: 10.1371 / tidskrift.pone.0012897
PubMed Abstrakt / CrossRef fulltext
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P. och Hayflick, S. J. (1993). i PLA2G6-associerad Neurodegeneration, eds M. P. Adam, H. H. Ardinger och Ra Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). CNS-involvering i Fabrys sjukdom: kliniska studier och bildstudier före och efter 12 månaders enzymersättningsterapi. J. Ärva. Metab. Dis. 27, 229–240. doi:10.1023/B: BOLI.0000028794.04349.91
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., snarare, M. I., Bhat, V., Gopinath, S., et al. (2016). Genetisk analys av PLA2G6 i 22 indiska familjer med infantil neuroaxonal dystrofi, atypisk sen debut neuroaxonal dystrofi och dystoni parkinsonism komplex. PLoS En 11: e0155605. doi: 10.1371 / tidskrift.pone.0155605
PubMed Abstrakt / CrossRef fulltext / Google Scholar
LaFountaine, JS, Fathe, K. och Smyth, H. D. (2015). Leverans och terapeutiska tillämpningar av genredigeringsteknik ZFNs. TALENs och CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016/j.ijpharm.2015.08.029
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Leke, M., Karczewski, K., Minikel, E., Samocha, K. E., banker, E., Fennell, T., et al. (2016). Analys av proteinkodande genetisk variation hos 60 706 människor. Natur 536, 285-291. doi: 10.1038 / nature19057
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Mänsklig genetisk variation förändrar CRISPR-Cas9 on-och off-targeting specificitet vid terapeutiskt implicerade loci. Proc. Natl. Acad. Sci. USA 14, E11257-E11266. doi: 10.1073/pnas.1714640114
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Levi, S. och Finazzi, D. (2014). Neurodegeneration med ackumulering av hjärnjärn: uppdatering om patogena mekanismer. Front. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef fulltext / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Mitokondriell lokalisering av cyklooxygenas – 2 och kalciumoberoende fosfolipas A2 i humana cancerceller: implikation i apoptosresistens. Exp. Cell Res. 306, 75-84. doi: 10.1016 / j. yexcr.2005.01.011
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Macauley, S. L. och Sands, M. S. (2009). Lovande CNS-riktad enzymersättningsterapi för lysosomala lagringssjukdomar. Exp. Neurol. 218, 5–8. doi: 10.1016/j.expneurol.2009.03.040
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Mehnaaz, L. (2016). Neurodegeneration med hjärnjärnackumulering (NBIA) tidigare Hallervorden – Spatz sjukdom. J. Assoc. Phys. Indien 64:132.
Google Scholar
han är en av de mest kända och mest kända i världen. (2006). PLA2G6, som kodar för ett fosfolipas A2, muteras i neurodegenerativa störningar med högt hjärnjärn. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Murphy, M. P. (2009). Hur mitokondrier producerar reaktiva syrearter. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., capit Kambodo, C. och Faria, A. (2016). Neurodegeneration med hjärnjärnackumulering: en fallrapport. Dement. Neuropsychol. 10, 160–164. doi: 10.1590 / S1980-5764-2016dn1002014
PubMed Abstrakt | CrossRef fulltext | Google Scholar
Nishiyama, J., Mikuni, T. och Yasuda, R. (2017). Virusmedierad genomredigering via homologistyrd reparation i mitotiska och postmitotiska celler i däggdjurshjärna. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j. neuron.2017.10.004
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F. och Farooqui, AA (2005). Distribution av kalciumoberoende fosfolipas A2 (iPLA2) i APA hjärnan. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Pardridge, W. M. (2002). Läkemedels-och geninriktning mot hjärnan med molekylära trojanska hästar. Nat. Rev. Drog Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Pardridge, W. M. (2005a). Molekylärbiologi av blod-hjärnbarriären. Mol. Bioteknol. 30, 57–70. doi: 10.1385 / MB:30:1:057
CrossRef fulltext / Google Scholar
Pardridge, W. M. (2005b). Blod-hjärnbarriären: flaskhals i hjärnans läkemedelsutveckling. NeuroRx 2, 3-14.
Google Scholar
han är en av de mest kända och mest kända i världen. (2015). Kalciumoberoende fosfolipaser A2 och deras roller i biologiska processer och sjukdomar. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Roca, C., Motas, S., Marc Ukrainian, S., Ribera, A., S Saborinchez, V., s Saborinchez, X., et al. (2017). Sjukdomskorrigering genom AAV-medierad genterapi i en ny musmodell av mukopolysackaridos typ IIID. Brum. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg / ddx058
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Scott, D. A. och Zhang, F. (2017). Konsekvenser av mänsklig genetisk variation i CRISPR-baserad terapeutisk genomredigering. Nat. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
K., Zhao, C., Zhang, X. H., Song, K. och Ma, Z. A. (2006). Kalciumoberoende fosfolipas A2 lokaliserar i och skyddar mitokondrier under apoptotisk induktion av staurosporin. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Smith, aj, Bainbridge, jw och Ali, rr (2012). Gentillskottsterapi för recessiva former av ärftliga retinala dystrofier. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Sorrentino, N. C., D ’ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Ett mycket utsöndrat sulfamidas konstruerat för att korsa blod-hjärnbarriären korrigerar hjärnskador hos möss med mukopolysackaridoser typ IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002/emmm.201202083
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Sorrentino, NC och Fraldi, A. (2016). Hjärnan inriktning i MPS-IIIA. Pediatr. Endocrinol. Rev. 13 (Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J. och Jones, SS (1997). Ett nytt cytosoliskt kalciumoberoende fosfolipas A2 innehåller åtta ankyrinmotiv. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstrakt / CrossRef fulltext / Google Scholar
amerikanska Food and Drug Administration (2017). FDA godkänner första behandlingen för en Form av Batten sjukdom. Finns på: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D. och Gottlieb, R. A. (2002). Hämning av mitokondriellt kalciumoberoende fosfolipas A2 (iPLA2) dämpar mitokondriell fosfolipidförlust och är hjärtskyddande. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Yu-Wai-Man, P. (2016). Genetisk manipulation för ärftliga neurodegenerativa sjukdomar: myt eller verklighet? Br. J. Oftalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., sång, K., et al. (2010). Skydd av betaceller i bukspottkörteln per grupp via fosfolipas A (2) – medierad reparation av mitokondriell membranperoxidation. Endokrinologi 151, 3038-3048. doi: 10.1210 / sv.2010-0016
PubMed Abstrakt / CrossRef Fulltext / Google Scholar