introductie
infantiele neuroaxonale dystrofie (Inad) is een autosomaal recessieve zeldzame neurodegeneratieve ziekte met onbekende frequentie. De symptomen beginnen meestal tussen de leeftijd van 6 maanden en 3 jaar. Voorafgaand aan die tijd ontwikkelen baby ‘ s zich normaal. Het eerste symptoom van INAD kan het vertragen van het bereiken van normale ontwikkelingsmijlpalen of regressie in ontwikkelingsmijlpalen zijn (Ramanadham et al., 2015). Hypotonie van de romp, scheelzien en nystagmus zijn vroege symptomen van de ziekte (Gregory et al., 2017). De progressie van de ziekte is snel en naarmate het vordert, meer verworven vaardigheden verloren. Spieren worden al snel hypotoon en later spastisch (Levi and Finazzi, 2014). Uiteindelijk gaat alle vrijwillige spiercontrole verloren. Spierzwakte kan ook leiden tot problemen bij het voeden en ademen. Naast nystagmus ervaren sommige kinderen visusverlies. Cognitieve functies gaan geleidelijk verloren en dementie ontwikkelt zich. De levensduur is over het algemeen 5 tot 10 jaar (Gregory et al., 1993; Jardim et al., 2004; Macauley And Sands, 2009).
moleculaire pathologie
infantiele Neuroaxonale dystrofie behoort tot een familie van neurodegeneratieve aandoeningen waaronder atypische laat-verworven neuroaxonale dystrofie (ANAD) en dystonie-Parkinsonismecomplex (DPC). De meeste gevallen van INAD worden geassocieerd met homozygote of samengestelde heterozygote mutaties in het PLA2G6-gen die de katalytische activiteit van het eiwitproduct beïnvloeden (Engel et al., 2010). Het pla2g6 gen codeert een groep via calcium-onafhankelijk fosfolipase A2 proteïne (PLA2G6 of iPLA2ß, 85 85/88 kDa) met een lipase en zeven ankyrine repeat-bevattende domeinen (Tang et al., 1997). PLA2G6 hydrolyseert de SN-2 acylketen van fosfolipiden en genereert vrije vetzuren en lysofosfolipiden.
fosfolipiden in het binnenste membraan van de mitochondriën zijn rijk aan onverzadigde vetzuren in de SN-2 Positie, in het bijzonder cardiolipine (Seleznev et al., 2006). Deze onverzadigde vetzuren zijn bijzonder kwetsbaar voor de overvloedige reactieve zuurstofspecies geproduceerd door de mitochondriën (Murphy, 2009) resulterend in peroxidized phospholipids in het binnenste membraan van de mitochondriën. PLA2G6 lokaliseert aan de mitochondriën (Williams en Gottlieb, 2002; Liou et al., 2005) consistent met een verhoogde vraag naar hydrolyse van de peroxideerde vetzuren in de SN-2 positie van fosfolipiden, wat leidt tot hermodeled fosfolipiden (Balsinde et al., 1995; Zhao et al., 2010). Wanneer PLA2G6 defect is, wordt de mitochondriën binnenmembraan integriteit beschadigd. PLA2G6 lokaliseert ook naar het axon (Ong et al., 2005; Seleznev et al., 2006) die op een verhoogde gelokaliseerde vraag voor phospholipid het remodelleren daar ook wijzen. De manifestatie van een dergelijke accumulatie in de hersenen is uniek voor belangrijke hersengebieden, zoals de basale ganglia die resulteerde in verschillende namen voor dezelfde onderliggende moleculaire pathogenese waarbij PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
ultrastructure analyse van neuronen in PLA2G6 knockout muizen is consistent met deze moleculaire pathologie. Mitochondriën met vertakte en tubulaire cristae, mitochondriën met gedegenereerde cristae, axonen met cytoskelet collaps, en partieel membraanverlies bij Axon terminals zijn waargenomen (Beck et al., 2011). Op microscopisch niveau verschijnen deze kenmerken als axonale zwellingen en sferoïde lichamen in pre-synaptische terminals (figuur 1) in het centrale of perifere zenuwstelsel.
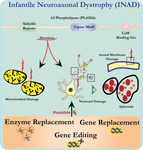
figuur 1. Infantiele neuroaxonale dystrofie (inad) is een neurodegeneratieve aandoening die verband houdt met mutaties in het pla2g6-gen. Verschillende mutaties van PLA2G6 leiden tot disfunctionele A2 fosfolipase die leidt tot mitochondriale en axonale membraandefecten. Deze tekorten veroorzaken neuronale schade gevisualiseerd als axonale zwellingen en accumulatie van pre-synaptische sferoïden. Enzymvervanging om functies te herstellen, genvervanging of bewerking om de defecte PLA2G6 te corrigeren zijn voorgestelde therapeutische strategieën.
Moleculaire Diagnostiek en Empowerment van patiënten met zeldzame ziekten
afgezien van specifieke klinische, elektrofysiologische en beeldvormende kenmerken, voorafgaand aan de beschikbaarheid van de volgende generatie sequencing, waren huidbiopten die axonale zwellingen en sferoïde lichamen in pre-synaptische terminals in het centrale of perifere zenuwstelsel de diagnostische criteria voor de bevestiging van INAD (Gregory et al., 2017; Jodice et al., 2017). Vaak waren meerdere biopten nodig om de diagnose te bevestigen. Gezinnen wachtten over het algemeen vele jaren op een diagnose. Met de dalende kosten van gen en genoom sequencing, beschikbaarheid van gerichte gen panel testen met diagnostische laboratoria voor niet-gediagnosticeerde neurologische ziekten, en het toenemende bewustzijn van artsen van de beschikbaarheid van genetische diagnostiek, families ontvangen diagnose sneller; soms binnen een jaar na het eerste symptoom verschijnen. De kinderen in deze families zijn nog jong en de families zijn gemotiveerd om samen met wetenschappers een behandeling voor de ziekte van hun kinderen te vinden. Om het onderzoeksproces te financieren, vormde een groep ouders van inad-patiënten de Inadcure Foundation. De stichting heeft aanzienlijke middelen voor onderzoek bijeengebracht en werkt samen met Rare Genomics Institute om hen te begeleiden bij het toekennen van onderzoeksbeurzen. Interessant, Een 2016 genetische analyse van 22 Indiase families met INAD, ANAD, en DPC gevonden dat 10/22 families (45.45%) ontbrak mutaties in de pla2g6 gen codering gebied (Kapoor et al., 2016). Het niet identificeren van schadelijke mutaties in het codeergebied van PLA2G6 benadrukt dat toekomstige moleculaire diagnostische inspanningen hele gensequencing nodig zouden hebben om mutaties in de intronische en regulerende gebieden van het pla2g6 gen bij patiënten met INAD te identificeren. Bovendien suggereert AD gevallen waarin PLA2G6 geassocieerde mutaties niet worden waargenomen dat de oorzaak van de ziekte te wijten zou kunnen zijn aan mutatie in andere genen dan PLA2G, die moet worden onderzocht.
mogelijke therapieën
de meeste therapieën die worden overwogen voor een zeldzame ziekte zoals INAD waarbij een defect enzym betrokken is, zijn enzymvervanging, genvervanging of gencorrectie. Wanneer een enzymdeficiëntie wordt veroorzaakt door een recessief genetisch defect, wordt aangenomen dat enzymvervanging of-suppletie het probleem kan verhelpen (Smith et al., 2012; Yu-Wai-Man, 2016). Experimenten om te bewijzen dat therapieën die het juiste gen of enzym leveren het inad fenotype zullen redden, moeten echter nog worden uitgevoerd en getest.
enzymvervangingstherapie (ert)
aangezien de hersenen het primaire aangetaste orgaan zijn bij INAD, zou de enzymvervangingstherapie voor inad hoogstwaarschijnlijk een infusie van het enzym in de hersenen vereisen. In 2017, ontving Biomarin FDA goedkeuring voor tripeptidylpeptidase 1 (cerliponase alfa) als behandeling voor de onderliggende oorzaak van latten ziekte, de deficiëntie van tripeptidylpeptidase (TPP1) een lysosomal enzym (V. S. Food Drug Administration, 2017). Tripeptidylpeptidase 1 (cerliponase alfa; 59 59 kDa) is de eerste ERT die direct wordt toegediend in de cerebrospinale vloeistof (CSF) van de hersenen. Andere ERT-geneesmiddelen die in de CSF kunnen worden toegediend, zijn in klinische studies (Jardim et al., 2004; Macauley And Sands, 2009).
de werkzaamheid van tripeptidylpeptidase 1 (cerliponase alfa) op het loopvermogen van patiënten met de ziekte van latten toont aan dat enzymvervangingstherapie voor ziekten die de hersenen aantasten theoretisch mogelijk is. Het Targeting van het vervangingsenzym in de mitochondriën voor inad zal een grotere uitdaging vormen dan het targeting van lysosomale enzymen, die al lange tijd succesvol was met IV ERT, zoals in het geval van de ziekte van Gaucher. Daarom zijn er nog steeds veel biologische problemen die specifiek zijn voor INAD die moeten worden opgelost:
• heeft de therapie voor inad mechanismen nodig die mogelijk PLA2G6 in de cel kunnen transporteren?
* welk mechanisme kan worden gebruikt om PLA2G6 mogelijk te lokaliseren tot de mitochondriën?
* is dat voldoende of moet PLA2G6 ook worden gelokaliseerd in het axon en zo ja, hoe?
* is er een speciale noodzaak om de doelgerichtheid van het perifere zenuwstelsel aan te pakken?
• kan een gedeeltelijke PLA2G6 de deficiëntie ervan redden of is een volledig eiwit nodig?
* Wat is een geschikt oplosmiddel voor toediening van PLA2G6 genproduct in de CSF?
* als het PLA2G6-enzym niet 100% zuiver is, wat zijn dan de contaminanten en zijn ze veilig?
• zijn er gegeneraliseerde intravasculaire complicaties als gevolg van PLA2G6-enzyminfusie in het CZS? Heeft de infusiedosis enige relevantie voor dergelijke complicaties?
een bijkomend probleem is dat de enzymvervangingstherapie de eerste plaatsing van een intracerebroventriculaire katheter en frequente infusies vereist. De plaatsing van de katheter vereist anesthesie en de infusies kunnen anesthesie afhankelijk van de samenwerking van de patiënten vereisen. Het aanpakken van de zorgen van pediatrische neurologen en ouders van kinderen met INAD met betrekking tot het gebruik van anesthesie op inad-patiënten met de juiste informatie zal verdere inspanning vereisen.
gentherapie/Genvervanging
het humane PLA2G-gen is ongeveer 70 kb met 17 exonen en 2 alternatieve exonen. De langste eiwitcodering transcript, echter, is slechts 3.3 kb en de eiwitcodering sequentie is iets meer dan 2,4 kb, die gemakkelijk kan worden verpakt in een virale vector lading. We zullen eerst een andere lysosomale opslagstoornis in het CNS onderzoeken die preklinische gegevens hebben die dan met INAD moeten worden vergeleken. Het gebrek aan preklinische gegevens van ERT in INAD is een significante kenniskloof in het veld, daarom is het cruciaal dat we andere preklinische modellen van CZS-aandoeningen observeren die gentherapie-studies ondergaan. Eerder, een preklinische studie bij muizen gericht gentherapie voor de lysosomale opslagstoornis mucopolysaccharidoses type IIIA (MPS-IIIA) door het verpakken van de juiste versie van het sulfamidase gen in een virale vector (Sorrentino et al., 2013). De studie maakte gebruik van het groeiende begrip van de bloed-hersenbarrière (BBB) die actief het transport van grote moleculen van bloed naar CNS reguleert door een proces genaamd transcytose (Pardridge, 2005b; Sorrentino and Fraldi, 2016).
Transcytose impliceert endocytose van liganden aan de luminale zijde, gemedieerd door ligand-specifieke receptoren (bijv. insulinereceptor, transferrinereceptor, en low-density-lipoproteïnereceptor, enz.,) verrijkt op het capillaire endotheel (Pardridge, 2005a). Dit wordt gevolgd door beweging van endocytose lading door het endotheel cytoplasma en uiteindelijk exocytose aan de abluminal (hersenen) kant, waardoor effectief het leveren van de lading over BBB (Pardridge, 2002). De preklinische studie MPS-IIIA produceerde een chimerisch sulfamidase-eiwit dat een BBB-bindend domein van apolipoproteïne B bevatte om opname door het endotheel te vergemakkelijken en ook een signaalpeptide van iduronaat-2-sulfatase om een efficiënte exocytose naar de abluminale kant van de BBB te helpen (Sorrentino et al., 2013). Een virale vector lading die codeert een chimerische sulfamidase werd vervolgens geladen op een adeno-geassocieerd virus (AAV) serotype 2/8 gericht op de lever (Sorrentino en Fraldi, 2016). Zo diende de lever als een interne fabriek die een constante toevoer van chimerisch-sulfamidase verstrekte, die in een 10-15% verhoging van hersenensulfamidase activiteit zelfs 7 maanden na lever gentherapie resulteerde. Deze toename van de activiteit van hersensulfamidase leidde tot kwantificeerbare verbetering van hersenpathologie en gedrag in het muismodel van MPS-IIIA (Sorrentino et al., 2013).
indien een soortgelijke studie voor INAD wordt uitgevoerd, zou deze een aantal kritische vragen over enzymvervanging voor inad-therapie beantwoorden. Als alternatief kan intra-vasculaire of intra-CSF toediening van op AAV9 gebaseerde gentherapie direct gericht zijn op het CZS (Bey et al., 2017; Roca et al., 2017). Nochtans, verstrekken de veelvoudige veranderingen die de patiënten INAD op hun gen PLAG26 hebben unieke uitdagingen aan gentherapie. Hoewel de grootte van het pla2g6 gen van 2-3 kb geen probleem zou moeten vormen voor zijn insertie in een virale vector, zijn de regelgevende complicaties van het corrigeren van PLA2G6 moeilijk te voorspellen. De regelgevende complicaties worden oncontroleerbaar vooral als de gentherapie niet goed binnen de doelweefsels kan worden gelokaliseerd. Enzymvervangingstherapie kan ook een probleem zijn als sommige mutantproducten dominant negatief blijken te zijn voor het wild-type PLAG26.
de meeste uitdagingen van mogelijke therapieën voor INAD komen voort uit het feit dat het een zeer zeldzame ziekte is. Nochtans, verstrekt het feit dat INAD een bekende genetische etiologie heeft wegen voor mogelijke therapie. Bovendien krijgt elke succesvolle therapie voor INAD de status van weesgeneesmiddel en alle bescherming die het krijgt omdat INAD een zeldzame ziekte is. Ondanks alle bovengenoemde uitdagingen vormt de status van weesgeneesmiddel dus een sterke stimulans voor onderzoekers van zeldzame ziekten en de biotechnologische industrie, om nog maar te zwijgen van het feit dat de genetische oorzaak al bekend is.
Gen / Base Editing
vanaf 2017 zijn ten minste 277 missense mutaties waargenomen in het humane PLA2G6 gen (Lek et al., 2016). Slechts een klein deel van PLA2G6 mutaties omvat frame verschuivingen, indels, nonsense mutaties en mutaties in de splice sites (Morgan et al., 2006). Zo is het corrigeren van het gen in de populatie van de doelcel of het corrigeren van de gemuteerde basissen van DNA een aantrekkelijke therapeutische weg. In de loop der jaren, zijn verscheidene hulpmiddelen ontwikkeld voor Gen het uitgeven, en het op CRISPR/Cas9-gebaseerde genoom het uitgeven wordt geprezen als baanbrekende technologie met een klinische proef die in 2018 wordt gepland. Deze technologieën gebruiken een bindende proteã ne van DNA die de bundel op een gespecificeerde manier ook kan splijten om ruimte voor de toevoeging van een nieuwe opeenvolging van DNA of correctie van de schadelijke basis van DNA te maken (LaFountaine et al., 2015). De technologie CRISPR / Cas9 heeft reeds verbazende therapeutische resultaten in verscheidene preklinische ziektemodellen (b.v., Duchene spierdystrofie, lever metabolische ziekten, enz. opgeleverd.,) (Dai et al., 2016). Bovendien, een groeiend begrip van de menselijke variatie stelt nieuwere uitdagingen voor gentherapie en het aandrijven van innovatie in de richting van een echte personalisatie van gen editing (Lessard et al., 2017; Scott and Zhang, 2017). Nieuwe technologieën zoals vSLENDR, een AAV virus, en CRISPR / Cas9-gemedieerde technologie om defecte genen in neuronen en andere zenuwstelselcellen te vervangen duwen gen-editing technologieën naar nieuwe grenzen (Nishiyama et al., 2017).
conclusie
infantiele Neuroaxonale dystrofie is een ernstige neurodegeneratieve ziekte met een bepaalde morbiditeit en mortaliteit. Deze zeldzame ziekte biedt een opwindende kans om beschikbare modi van de volgende generatie therapie te herwaarderen en nieuwere te genereren. De opkomende Congruentie op de klinische diagnostische criteria voor INAD zal naar verwachting een impuls geven aan verbeterde moleculaire diagnostiek. Deze vooruitgang zal ons naar verwachting leiden tot het ontwikkelen van betaalbare therapieën die kwantificeerbare verbetering van de kwaliteit van leven van INAD-patiënten zouden bieden en de progressie van de ziekte zouden vertragen of verbeteren. We hebben een aantal succesverhalen in de ziekte van latten en de mucopolysaccharidoses die ons voorzien van de inspiratie om de juiste vragen te stellen om inad therapie een realiteit. Bovendien zouden de groeiende virale en niet-virale benaderingen voor CRISPR/Cas9 gebaseerde gen het uitgeven ook nieuwere therapeutische wegen voor INAD moeten openen.
Auteursbijdragen
PB en FA voorbereid voorontwerp. SR en AC bewerkt en toegevoegd meer secties aan het manuscript. DF, AP en LP redigeerden het manuscript.
verklaring inzake belangenconflicten
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., en Dennis, E. A. (1995). De remming van calcium-onafhankelijke phospholipase A2 verhindert arachidonic zure opname en phospholipid remodellering in p388d1 macrofagen. Proc. Natl. Acad. Sci. U. S. A. 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Efficiënte CNS targeting in volwassen muizen door intrathecale infusie van single-stranded AAV9-GFP voor gentherapie van neurologische wanorde. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstract / CrossRef Full Text / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 voor in vivo gentherapie: belofte en hindernissen. Mol. Ther. Nucleïnezuren 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstract / CrossRef Full Text / Google Scholar
Engel, L. A., Jing, Z., O’ Brien, D. E., Sun, M., and Kotzbauer, P. T. (2010). De katalytische functie van PLA2G6 wordt verstoord door mutaties geassocieerd met infantiele neuroaxonale dystrofie, maar niet door dystonie-parkinsonisme. PLoS One 5: e12897. doi: 10.1371 / journal.pone.0012897
PubMed Abstract / CrossRef volledige tekst
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (1993). in PLA2G6-Associated Neurodegeneration, eds M. P. Adam, H. H. Ardinger, and R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Betrokkenheid van het CZS bij de ziekte van Fabry: klinische en beeldvormende onderzoeken vóór en na 12 maanden enzymvervangingstherapie. J. Erven. Metab. Dis. 27, 229–240. doi: 10.1023 / B: BOLI.0000028794.04349.91
PubMed Abstract / CrossRef Full Text / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Rather, M. I., Bhat, V., Gopinath, S., et al. (2016). Genetische analyse van PLA2G6 in 22 Indiase families met infantiele neuroaxonale dystrofie, atypische late-onset neuroaxonale dystrofie en dystonie parkinsonisme complex. PLoS One 11: e0155605. doi: 10.1371 / journal.pone.0155605
PubMed Abstract / CrossRef Full Text / Google Scholar
LaFountaine, J. S., Fathe, K., en Smyth, H. D. (2015). Levering en therapeutische toepassingen van gen editing technologies ZFNs. TALENs, en CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed Abstract / CrossRef Full Text / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analyse van eiwitcoderende genetische variatie bij 60.706 mensen. Nature 536, 285-291. doi: 10.1038 / nature19057
PubMed Abstract / CrossRef Full Text / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Menselijke genetische variatie verandert CRISPR-Cas9 on-en off-targeting specificiteit bij therapeutisch betrokken loci. Proc. Natl. Acad. Sci. U. S. A. 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
PubMed Abstract / CrossRef Full Text / Google Scholar
Levi, S., and Finazzi, D. (2014). Neurodegeneratie met ijzerophoping in de hersenen: update over pathogene mechanismen. Voorkant. Farmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Full Text / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Mitochondriale lokalisatie van cyclo-oxygenase-2 en calcium-onafhankelijke fosfolipase A2 in menselijke kankercellen: implicatie in apoptose resistentie. Exp. Cel Res. 306, 75-84. doi: 10.1016 / j.yexcr.2005.01.011
PubMed Abstract / CrossRef Full Text / Google Scholar
Macauley, S. L., And Sands, M. S. (2009). Veelbelovende CZS-gerichte enzymvervangingstherapie voor lysosomale opslagziekten. Exp. Neurol. 218, 5–8. doi: 10.1016 / j.expneurol.2009.03.040
PubMed Abstract / CrossRef Full Text / Google Scholar
Mehnaaz, L. (2016). Neurodegeneratie met hersenijzeraccumulatie (NBIA) voorheen ziekte van Hallervorden-Spatz. J. Assoc. Phys. India 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K., Morton, J. E. V., Gregory, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, codeert een fosfolipase A2, wordt gemuteerd in neurodegeneratieve aandoeningen met hoog hersenijzer. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed Abstract / CrossRef Full Text / Google Scholar
Murphy, M. P. (2009). Hoe mitochondriën reactieve zuurstofsoorten produceren. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstract / CrossRef Full Text / Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C., en Faria, A. (2016). Neurodegeneratie met hersenijzerophoping: een case report. Dement. Neuropsychol. 10, 160–164. doi: 10.1590 / S1980-5764-2016DN1002014
PubMed Abstract / CrossRef Full Text / Google Scholar
Nishiyama, J., Mikuni, T., and Yasuda, R. (2017). Virus-gemedieerde genoom het uitgeven via homologie-gerichte reparatie in mitotic en postmitotic cellen in zoogdierhersenen. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j.neuron.2017.10.004
PubMed Abstract / CrossRef Full Text / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., and Farooqui, A. A. (2005). Distributie van calcium-onafhankelijke fosfolipase A2 (iPLA2) in apenhersenen. J. Neurocytol. 34, 447–458. doi: 10.1007/s11068-006-8730-4
PubMed Abstract / CrossRef Full Text / Google Scholar
Pardridge, W. M. (2002). Drug en Gen gericht op de hersenen met moleculaire Trojaanse paarden. Nat. Rev. Drug Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed Abstract / CrossRef Full Text / Google Scholar
Pardridge, W. M. (2005a). Moleculaire biologie van de bloed-hersenbarrière. Mol. Biotechnol. 30, 57–70. doi: 10.1385/MB:30: 1:057
CrossRef Full Text / Google Scholar
Pardridge, W. M. (2005b). De bloed-hersenbarrière: bottleneck in brain drug development. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Calcium-onafhankelijke fosfolipasen A2 en hun rol in biologische processen en ziekten. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstract / CrossRef Full Text / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Ziektecorrectie door AAV-gemedieerde gentherapie in een nieuw muismodel van mucopolysaccharidose type IIID. Brom. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg / ddx058
PubMed Abstract / CrossRef Full Text / Google Scholar
Scott, D. A., and Zhang, F. (2017). Implicaties van menselijke genetische variatie in CRISPR-gebaseerd therapeutisch genoom het uitgeven. Nat. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstract / CrossRef Full Text / Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K., and Ma, Z. A. (2006). Calcium-onafhankelijke fosfolipase A2 lokaliseert in en beschermt mitochondriën tijdens apoptotische inductie door staurosporine. J. Biol. Scheikunde. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstract / CrossRef Full Text / Google Scholar
Smith, A. J., Bainbridge, J. W., and Ali, R. R. (2012). Gensuppletietherapie voor recessieve vormen van erfelijke retinale dystrofie. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstract / CrossRef Full Text / Google Scholar
Sorrentino, N. C., D ‘ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Een hoog afgescheiden sulfamidase die wordt ontworpen om de bloed-hersenbarrière te kruisen corrigeert hersenletsels van muizen met mucopolysaccharidoses type IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed Abstract / CrossRef Full Text / Google Scholar
Sorrentino, N. C., en Fraldi, A. (2016). Doel van de hersenen in MPS-IIIA. Kinderarts. Endocrinol. Rev.13(Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., and Jones, S. S. (1997). Een nieuwe cytosolic calcium-onafhankelijke fosfolipase A2 bevat acht ankyrine motieven. J. Biol. Scheikunde. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstract / CrossRef Full Text / Google Scholar
U. S. Food and Drug Administration (2017). FDA keurt eerste behandeling voor een vorm van latten ziekte. Beschikbaar op: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., and Gottlieb, R. A. (2002). Remming van mitochondriale calcium-onafhankelijke fosfolipase A2 (iPLA2) verzwakt mitochondriale fosfolipideverlies en is cardioprotectief. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstract / CrossRef Full Text / Google Scholar
Yu-Wai-Man, P. (2016). Genetische manipulatie voor erfelijke neurodegeneratieve ziekten: mythe of realiteit? Br. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed Abstract / CrossRef Full Text / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et al. (2010). Bescherming van pancreatische bètacellen door groep via fosfolipase A (2) – gemedieerde reparatie van mitochondriale membraanperoxidatie. Endocrinologie 151, 3038-3048. doi: 10.1210/nl.2010-0016
PubMed Abstract / CrossRef Full Text / Google Scholar