Introduction
Infantile neuroaxonal dystrophy(INAD)は、頻度が不明な常染色体劣性のまれな神経変性疾患です。 徴候の手始めは6か月と3歳の間に一般に起こります。 その前に、幼児は正常に発達する。 INADの最初の症状は、正常な発達マイルストーンの達成の減速または発達マイルストーンの回帰であり得る(Ramanadham et al., 2015). 体幹低血圧、斜視、および眼振は、この疾患の初期症状である(Gregory et al., 2017). 病気の進行は急速であり、進行するにつれて、より多くの獲得されたスキルが失われる。 筋肉はすぐに低張性になり、後に痙性になる(Levi and Finazzi、2014)。 最終的には、すべての自発的な筋肉の制御が失われます。 筋肉の衰弱はまた、摂食および呼吸の困難につながる可能性がある。 眼振に加えて、何人かの子供は視力喪失を経験する。 認知機能は徐々に失われ、認知症が発症する。 寿命は一般に5〜1 0年である(Gregory e t a l. ら、1 9 9 3;Jardim e t a l. ら,2 0 0 4;Macouley and Sands,2 0 0 9)。
分子病理学
小児神経軸索ジストロフィーは、非定型遅発性神経軸索ジストロフィー(ANAD)およびジストニアパーキンソニズム複合体(DPC)を含む神経変性障害のファミリーに属している。 INADのほとんどの症例は、そのタンパク質産物の触媒活性に影響を及ぼすPLA2G6遺伝子におけるホモ接合または化合物ヘテロ接合変異と関連している(Engel et al., 2010). PL a2g6遺伝子は、カルシウム非依存性ホスホリパーゼA2タンパク質(PL A2G6またはIPL A2β、≧8 5/8 8kDa)を介して、リパーゼおよび7つのアンキリンリピート, 1997). PLA2G6はリン脂質のsn-2アシル鎖を加水分解し、遊離脂肪酸およびリゾリン脂質を生成する。
ミトコンドリアの内膜中のリン脂質は、sn-2位の不飽和脂肪酸、特にカルジオリピンが豊富である(Seleznev et al., 2006). これらの不飽和脂肪酸は、ミトコンドリア(Murphy、2009)によって産生される豊富な活性酸素種に対して特に脆弱であり、その結果、ミトコンドリアの内膜に過酸 PL A2G6はミトコンドリアに局在する(Williams and Gottlieb,2 0 0 2;Liou e t a l. リン脂質のsn-2位における過酸化脂肪酸の加水分解に対する需要の増加と一致して(Balsinde et al.,2005)、リン脂質の再構築につながる(Balsinde et al.,2005)。 ら、1 9 9 5;Zhao e t a l., 2010). PLA2G6が不完全であるとき、mitochondriaの内部の膜の完全性は損なわれます。 PL A2G6はまた、軸索に局在化する(Ong e t a l. ら、2 0 0 5;Seleznev e t a l.,2006)は、リン脂質リモデリングの局所的な需要の増加を示している。 脳におけるそのような蓄積の発現は、大脳基底核のような主要な脳領域に特有のものであり、これは、PLAG2 6を含む同じ根底にある分子病因のための様々な名, 2016).
PLA2G6ノックアウトマウスにおけるニューロンの超微細構造解析は、この分子病理と一致しています。 分枝および管状クリスタを有するミトコンドリア、変性クリスタを有するミトコンドリア、細胞骨格崩壊を有する軸索、および軸索末端における部分的な膜損失が観察されている(Beck et al., 2011). 微視的なレベルでは、これらの特徴は、中枢または末梢神経系のシナプス前終末(図1)に軸索腫脹および回転楕円体として現れる。
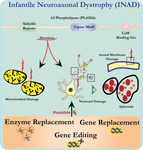
図1. 小児神経軸索ジストロフィー(INAD)は、PLA2G6遺伝子の変異に関連する神経変性疾患である。 PLA2G6のさまざまな突然変異はmitochondrialおよび軸索の膜の欠陥の原因となる機能不全のA2ホスホリパーゼに導きます。 これらの欠陥は、軸索の腫脹とシナプス前スフェロイドの蓄積として視覚化された神経損傷を引き起こす。 機能を回復するための酵素置換、欠陥のあるPLA2G6を修正するための遺伝子置換または編集が提案されている治療戦略である。
分子診断と希少疾患患者のエンパワーメント
特定の臨床的、電気生理学的、およびイメージング的特徴とは別に、次世代シーケンシングが利用可能になる前に、中枢または末梢神経系のシナプス前終末における軸索腫脹および回転楕円体を示す皮膚生検は、INADの確認のための診断基準であった(Gregory et al. ら、2 0 1 7;Iodice e t a l., 2017). 多くの場合、診断を確定するために複数の生検が必要であった。 家族は一般的に診断のために何年も待っていました。 遺伝子およびゲノム配列決定のコストの減少、診断されていない神経疾患のための診断ラボでの標的遺伝子パネル検査の利用可能性、および遺伝学的診断の利用可能性についての医師の意識の高まりに伴い、家族はより迅速に診断を受けている;時には最初の症状が現れてから一年以内に。 これらの家族の子供たちはまだ若く、家族は子供の病気の治療法を見つけるために科学者と提携するよう動機づけられています。 研究プロセスに資金を供給するために、INAD患者の両親のグループがINADcure財団を形成しました。 財団は、研究のためのかなりの資金を調達しており、研究助成金の授与にそれらを導くために希少ゲノミクス研究所と提携しています。 興味深いことに、INAD、ANAD、およびDPCを持つ22のインドの家族の2016年の遺伝子分析は、10/22家族(45.45%)がPLA2G6遺伝子コード領域に変異を欠いていたことがわか, 2016). PLA2G6のコード領域における有害な変異を識別するために失敗は、将来の分子診断の努力は、INAD影響を受けた患者におけるPLA2G6遺伝子のイントロニ さらに、PLA2G6関連変異が観察されないINAD症例は、疾患の原因がpla2G以外の遺伝子の変異によるものであり、探索する必要があることを示唆している。
可能な治療法
欠陥のある酵素を含むINADのようなまれな疾患に対して考慮される治療法のほとんどは、酵素置換、遺伝子置換または遺伝子修正で 酵素欠乏症が劣性遺伝的欠損によって引き起こされる場合、酵素置換または補充が問題を修正することができると仮定される(Smith et al. 2012年、Yu-Wai-Man、2016年)。 しかし、正しい遺伝子または酵素を提供する治療法がINAD表現型を救助することを証明するための実験はまだ実施されておらず、試験されていない。
酵素補充療法(ERT)
脳はINADの影響を受ける主要な器官であるため、INADの酵素補充療法では脳への酵素の注入が必要になる可能性が最も高い。 バイオマリンは2017年に、バッテン病の根本的な原因であるリソソーム酵素であるトリペプチジルペプチダーゼ(TPP1)の欠乏に対する治療薬として、トリペプチジルペプチダーゼ1(cerliponase alfa)のFDA承認を受けました(米国食品医薬品局、2017)。 トリペプチジルペプチダーゼ1(セリポナーゼアルファ; 59kDa以下)は、脳の脳脊髄液(CSF)に直接投与される最初のERTである。 CSFに投与することができる他のERT薬は、臨床試験中である(Jardim e t a l. ら,2 0 0 4;Macouley and Sands,2 0 0 9)。
トリペプチジルペプチダーゼ1(cerliponase alfa)がバッテン病患者の歩行能力に及ぼす有効性は、脳に影響を与える疾患に対する酵素補充療法が理論的に可能であることを示している。 INADのためのミトコンドリアへの置換酵素の標的化は,Gaucher病の場合のように長い間IVERTによって既に成功していたリソソーム標的化よりも困難である。 したがって、INADに特有の多くの生物学的問題を解決する必要があります:
•INADの治療には、おそらくPLA2G6を細胞内に輸送できるメカニズムが必要
•PLA2G6をミトコンドリアに局在させるためにどのようなメカニズムを使用できますか?
•それで十分なのか、それともPLA2G6も軸索に局在する必要があるのか、もしそうなら、どうすればよいのでしょうか?
•末梢神経系の標的化に対処するための特別な必要性はありますか?
•部分的なPLA2G6はその欠乏を救うことができますか、それともタンパク質全体が必要ですか?
•PLA2G6遺伝子産物をCSFに送達するための適切な溶媒は何ですか?
•PLA2G6酵素が100%純粋でない場合、その汚染物質は何であり、安全ですか?
•CNSへのPLA2G6酵素注入による一般化された血管内合併症はありますか? 注入用量はそのような合併症との関連性がありますか?
追加の問題は、酵素補充療法では、脳室内カテーテルの初期配置と頻繁な注入が必要であるということです。 カテーテルの配置は麻酔を要求し、注入は患者の協同によって麻酔を要求するかもしれません。 適切な情報を持つINAD患者に麻酔を使用することに関して、小児神経科医およびINADの子供の両親の懸念に対処するには、さらなる努力が必要です。
遺伝子治療/遺伝子置換
ヒトPLA2G遺伝子は約70kbで、17個のエクソンと2個の代替エクソンがある。 しかし、最長のタンパク質コード転写物はわずか3.3kbであり、タンパク質コード配列はわずか2.4kbを超えており、ウイルスベクターカーゴに容易にパッケージ化することができる。 我々は、最初にINADと比較する前臨床データを持っているCNSにおける別のリソソーム貯蔵障害を探索します。 INADにおけるERの前臨床データの欠如は、それが我々が遺伝子治療研究を受けているCNS疾患の他の前臨床モデルを観察することが重要である理由である、 以前に、マウスにおける前臨床研究では、ウイルスベクター内にスルファミダーゼ遺伝子の正しいバージョンを包装することにより、リソソーム貯蔵障害ムコ多糖症IIIA型(MPS−IIIA)の遺伝子治療に対処した(Sorrentino e t a l., 2013). この研究は、トランスシトーシスと呼ばれるプロセスによって血液から中枢神経系への大きな分子の輸送を積極的に調節する血液脳関門(BBB)の理解
トランスシトーシスは、リガンド特異的受容体(例えば、インスリン受容体、トランスフェリン受容体、低密度リポタンパク質受容体など)によって媒介される管腔側のリガンドのエンドサイトーシスを伴う。ら(Pardridge,2 0 0 5a)。 これに続いて、内皮細胞質を通ってエンドサイトーシスされた貨物が移動し、最終的に無管腔(脳)側でエキソサイトーシスが起こり、したがって効果的にBBBを横切って貨物を送達する(Pardridge、2002)。 MPS-IIIA前臨床研究では、内皮による取り込みを容易にするためにアポリポタンパク質BからBBB結合ドメインを含むキメラスルファミダーゼタンパク質と、BBBの無管側に向かって効率的なエキソサイトーシスを支援するためにイデュロン酸-2-スルファターゼからのシグナルペプチドを製造した(Sorrentino et al., 2013). キメラスルファミダーゼをコードするウイルスベクター貨物は、肝臓を標的とするアデノ随伴ウイルス(AAV)血清型2/8にロードされた(Sorrentino and Fraldi、2016)。 従ってレバーはレバー遺伝子療法の後の頭脳のスルファミダーゼの活動の10-15%増加で7か月起因したキメラスルファミダーゼの一定した供給を提供する内部工場として役立った。 脳スルファミダーゼ活性レベルのこの増加は、MPS−IIIAのマウスモデルにおける脳の病理学および行動転帰の定量化可能な改善をもたらした(Sorrentino e t a l., 2013).
同様の研究がINADに対して実施されれば、INAD療法のための酵素置換に関するいくつかの重要な質問に答えるだろう。 あるいは、AAV9ベースの遺伝子治療産物の血管内投与またはCSF内投与は、CNSを直接標的とすることができる(Bey e t a l. ら、2 0 1 7;Roca e t a l., 2017). しかし、INAD患者がPLAG26遺伝子に有する複数の変異は、遺伝子治療に固有の課題を提供する。 2-3kbのPLA2G6遺伝子のサイズは、ウイルスベクターへの挿入のための問題を提起すべきではないが、しかし、PLA2G6を修正することから調節合併症 特に遺伝子治療が標的組織内で適切に局在化することができない場合、調節合併症は制御不能になる。 いくつかの変異体生成物が野生型PLAG26に対して優性陰性であることが判明した場合、酵素補充療法も問題になる可能性がある26。
INADの可能な治療法の課題のほとんどは、それが超まれな病気であるという事実から来ています。 しかし、INADが既知の遺伝的病因を有するという事実は、可能な治療のための手段を提供する。 さらに、INADのためのどの巧妙な療法でもINADがまれな病気であるので孤児の薬剤の状態および得るすべての保護を受け取ります。 したがって、前述のすべての課題にもかかわらず、孤児薬の状態は、遺伝的原因がすでに知られているときはもちろんのこと、希少疾患研究者やバイオ
遺伝子/塩基編集
2017年現在、ヒトPLA2G6遺伝子に少なくとも277のミスセンス変異が観察されている(Lek et al., 2016). PL a2G6変異のほんの一部には、フレームシフト、インデル、ナンセンス変異、およびスプライス部位における変異が含まれる(Morgan e t a l., 2006). したがって、標的細胞集団中の遺伝子を修正するか、または変異したDNA塩基を修正することは、魅力的な治療手段である。 長年にわたり、遺伝子編集のためのいくつかのツールが開発されており、CRISPR/Cas9ベースのゲノム編集は、2018年に計画された臨床試験で画期的な技術として これらの技術は、新たなDNA配列の挿入または有害なDNA塩基の補正のための空間を作るために、特定の方法で鎖を切断することもできるDNA結合タン, 2015). CRISPR/Cas9技術は、すでにいくつかの前臨床疾患モデル(例えば、デュセン筋ジストロフィー、肝臓代謝疾患など)で驚異的な治療結果をもたらしています。,)(Dai et al., 2016). さらに、ヒトの変異の理解の高まりは、遺伝子治療に新たな課題を提起し、遺伝子編集の真のパーソナライゼーションに向けた革新を推進している(Lessard et al. ら、2017年;ScottおよびZhang、2017年)。 VSLENDR、A AVウイルス、および神経細胞および他の神経系細胞における欠損遺伝子を置換するCRISPR/Cas9媒介技術のような新技術は、遺伝子編集技術を新たなフロンティアに押し進めている(Nishyama e t a l., 2017).
結論
小児神経軸索ジストロフィーは、ある種の罹患率および死亡率を有する重度の神経変性疾患である。 このまれな病気は次世代療法の利用できるモードをrevalidateし、より新しい物を発生させる刺激的な機会を提供する。 INADの臨床診断基準に関する新たな一致は、強化された分子診断に向けた弾みを提供することが期待されています。 この進歩は、INAD患者の生活の質の定量化可能な改善を提供し、疾患の進行を遅らせるか、または改善する手頃な価格の治療法を開発するために私たち 私達はInad療法に現実をするために右の質問をするようにインスピレーションを私達に与える当て木の病気およびmucopolysaccharidosesのあるサクセス-ストーリーを強調した。 さらに、CRISPR/Cas9ベースの遺伝子編集のためのウイルスおよび非ウイルスのアプローチの成長はまた、INADのための新しい治療の道を開く必要があります。
著者の貢献
PBとFAは予備草案を準備しました。 SRとACは原稿に多くのセクションを編集し、追加しました。 DF、AP、LPが原稿を編集しました。
利益相反に関する声明
著者らは、本研究は、利益相反の可能性があると解釈される可能性のある商業的または財政的関係がない場合に行われた
Balsinde,J.,Bianco,I.D.,Ackermann,E.J.,Conde-Frieboes,K.,And Dennis,E.A.(1995). カルシウム非依存性ホスホリパーゼA2の阻害は、p388D1マクロファージにおけるアラキドン酸の取り込みとリン脂質リモデリングを防止します。 プロク… ナトル アカド サイ… 92,8527-8531 ドイ:10.1073/pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). 神経疾患の遺伝子治療のための一本鎖AAV9-GFPの髄腔内注入による成体マウスにおける効率的なCNSターゲティング。 ジーン-サー 24:325. 土井:10.1038/gt.2017.18
PubMed要約/CrossRef全文|Google Scholar
Dai,W.-J.,Zhu,L.-Y.,Yan,Z.-Y.,Xu,Y.,Wang,Q.-L.,Lu,X.-J.,et al. (2016). IN vivo遺伝子治療のためのCRISPR-Cas9:約束とハードル。 モル Ther。 核酸5:e3 4 9. ドイ:10.1038/mtna.2016.58
PubMed要約/CrossRef全文/Google Scholar
Engel,L.A.,Jing,Z.,O’Brien,D.E. とができるようになりました。 PLA2G6の触媒機能は、小児神経軸索ジストロフィーではなく、ジストニア-パーキンソニズムに関連付けられている突然変異によって損なわれます。 PLoS One5:e12897. doi:10.1371/ジャーナル。ポネ0012897
PubMed要約/CrossRef全文
とができるようになることを期待しています。 A.Pagon,Seattle,W A:Genereviews.
グレゴリー,A.,Kurian,M.A.,Maher,E.R.,Hogarth,P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Fabryの病気のCNSの介入:酵素の取り替え療法の12か月の前後の臨床およびイメージ投射調査。 J.インシード。 メタブ Dis。 27, 229–240. 土井:10.1023/B:BOLI.0000028794.04349.91
PubMed要約/CrossRef全文/Google Scholar
Kapoor,S.,Shah,M.H.,Singh,N.,Rather,M.I.,Bhat,V.,Gopinath,S.,et al. (2016). 小児神経軸索ジストロフィー、非定型遅発性神経軸索ジストロフィーとジストニアパーキンソニズム複合体と22インドの家族におけるPLA2G6の遺伝学 PLoS One11:e0155605. doi:10.1371/ジャーナル。ポネ0155605
PubMed要約/CrossRef全文/Google Scholar
とができるようになりました。 遺伝子編集技術ZFNsの送達および治療応用。 TALENSおよびCRISPR/Cas9。 Int. J.ファーム 494, 180–194. 土井:10.1016/j.ijpharm.2015.08.029
PubMed要約/CrossRef全文/Google Scholar
Lek,M.,Karczewski,K.,Minikel,E.,Samocha,K.E.,Banks,E.,Fennell,T.,et al. (2016). 60,706人のヒトにおけるタンパク質コード遺伝的変異の分析。 自然536、285-291。 土井:10.1038/nature19057
PubMed要約/CrossRef全文/Google Scholar
Lessard,S.,Francioli,L.,Alfoldi,J.,Tardif,J.C.,Ellinor,P.T.,MacArthur,D.G.,et al. (2017). ヒトの遺伝的変異は、治療的に関与する遺伝子座でのCRISPR-Cas9オンおよびオフターゲティング特異性を変化させる。 プロク… ナトル アカド サイ… 米国14、E11257-E11266。 ドイ:10.1073/pnas.1714640114
PubMed要約/CrossRef全文/Google Scholar
とができるようになりました。 脳の鉄蓄積による神経変性: 病原性メカニズムに関する更新。 フロント ファーマコール… 5:99. ドイ:10.3389/fphar.2014.00099
CrossRef全文/Google Scholar
Liou,J.Y.,Aleksic,N.,Chen,S.F.,Han,T.J.,Shyue,S.K.,Wu,K.K.,et al. (2005). ヒト癌細胞におけるシクロオキシゲナーゼ-2およびカルシウム非依存性ホスホリパーゼA2のミトコンドリア局在:アポトーシス抵抗性の含意。 経験値 セルRes.306、75-84。 土井:10.1016/j.yexcr.2005.01.011
PubMed要約/CrossRef全文/Google Scholar
マコーリー,S.L.,とサンズ,M.S.(2009). リソソーム貯蔵疾患のための有望なCNS指向の酵素補充療法。 経験値 ニューロール 218, 5–8. ドイ:10.1016/j.expneurol.2009.03.040
PubMed抄録/クロスリファレンス全文/Google Scholar
Mehnaaz,L.(2016). 脳鉄蓄積(NBIA)を伴う神経変性以前はHallervorden-Spatz病であった。 J.Assoc. フィス 64:132
Morgan,N.V.,Westaway,S.K.,Morton,J.E.V.,Gregory,A.,Gissen,P.,Sonek,S.,et al. (2006). ホスホリパーゼA2をコードするPLA2G6は、高脳鉄と神経変性疾患で変異しています。 ナット ジュネット 38, 752–754. doi:10.1038/ng1826
PubMed要約/CrossRef全文/Google Scholar
マーフィー,M.P.(2009). ミトコンドリアが活性酸素をどのように産生するか。 バイオケム 417件中1-13件目 doi:10.1042/BJ20081386
PubMed要約/CrossRef全文/Google Scholar
ることができると考えています。 脳鉄蓄積による神経変性:症例報告。 デメント ニューロサイチョル… 10, 160–164. doi:10.1590/S1980-5764-2016DN1002014
PubMed要約/CrossRef全文/Google Scholar
ることを明らかにしました。 哺乳類の脳における有糸分裂および有糸分裂後の細胞における相同性指向の修復を介してウイルス媒介ゲノム編集。 96,755e5-768e5. 土井:10.1016/j.neuron.2017.10.004
PubMed要約/CrossRef全文/Google Scholar
ることができると考えられている。 サルの脳におけるカルシウム非依存性ホスホリパーゼA2(ipla2)の分布。 J.Neurocytol. 34, 447–458. 土井:10.1007/s11068-006-8730-4
PubMed Abstract|CrossRef全文|Google Scholar
パードリッジ,W.M.(2002). 分子トロイの木馬で脳を標的とする薬物および遺伝子。 ナット ドラッグ-ディスコブ… 1, 131–139. doi:10.1038/nrd725
PubMed要約/CrossRef全文/Google Scholar
Pardridge,W.M.(2005a). 血液脳関門の分子生物学。 モル バイオテクノール 30, 57–70. ドイ:10.1385/MB:30:1:057
クロスリファレンス全文/Google Scholar
Pardridge,W.M.(2005b). 血液脳関門:脳の医薬品開発におけるボトルネック。 新潟県新潟市中央区新発田2丁目3-14
Ramanadham,S.,Ali,T.,Ashley,J.W.,Bone,R.N.,Hancock,W.D.,Lei,X.,et al. (2015). カルシウム非依存性ホスホリパーゼA2と生物学的プロセスと疾患におけるその役割。 J.Lipid Res.56,1643-1668. ドイ:10.1194/jlr。R058701
PubMed要約/CrossRef全文/Google Scholar
Roca,C.,Motas,S.,Marcó,S.,Ribera,A.,Sánchez,V.,Sánchez,X.,et al. (2017). MucopolysaccharidosisのタイプIIIDの新しいマウスモデルのAAV仲介された遺伝子療法による病気の訂正。 ハム モル ジュネット 26, 1535–1551. doi:10.1093/hmg/ddx058
PubMed要約/CrossRef全文/Google Scholar
ることができます。 CRISPRベースの治療ゲノム編集におけるヒトの遺伝的変異の影響。 ナット メッド 23:1095. ドイ:10.1038/nm.4377
PubMed要約/CrossRef全文/Google Scholar
セレズネフ,K.,Zhao,C.,Zhang,X.H.、歌、K.、およびMa、Z.A.(2006年)。 カルシウム非依存性ホスホリパーゼA2に局在し、スタウロスポリンによるアポトーシス誘導中にミトコンドリアを保護します。 J.Biol. ケム 281, 22275–22288. ドイ:10.1074/jbc.M604330200
PubMed要約/CrossRef全文/Google Scholar
とができるようになることを期待しています。 遺伝性網膜ジストロフィーの劣性形態のための遺伝子補充療法。 ジーン-サー 19, 154–161. 土井:10.1038/gt.2011.161
PubMed要約/CrossRef全文/Google Scholar
ソレンティーノ C.,D’Orsi,L.,Sambri,I.,Nusco,E.,Monaco,C.,Spampanato,C.,et al. (2013). 血頭脳の障壁を交差させるために設計される非常に分泌されたスルファミダーゼはmucopolysaccharidosesのタイプIIIAが付いているマウスの頭脳の損害を訂正します。 エンボモル メッド 5, 675–690. ドイ:10.1002/emmm.201202083
PubMed要約/CrossRef全文/Google Scholar
とができるようになりました。 MPS-IIIAにおける脳ターゲティング。 小児科 エンデクリノール 第13回(第13回) 1), 630–638.
Tang,J.,Kriz,R.W.,Wolfman,N.,Shaffer,M. ら、seehra,J.,およびJones,S. 新規細胞質カルシウム非依存性ホスホリパーゼA2は、八アンキリンモチーフが含まれています。 J.Biol. ケム 272, 8567–8575. ドイ:10.1074/jbc.272.13.8567
PubMed要約|CrossRef全文|Google Scholar
米国食品医薬品局(2017)。 FDAは当て木の病気の形態のための最初処置を承認する。 で利用可能: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams,S.D.,And Gottlieb,R.A.(2002). ミトコンドリアカルシウム非依存性ホスホリパーゼA2(ipla2)の阻害は、ミトコンドリアのリン脂質の損失を減衰させ、心臓保護性である。 バイオケム J.362(Pt1),23-32. doi:10.1042/bj3620023
PubMed要約/CrossRef全文/Google Scholar
Yu-Wai-Man,P.(2016). 遺伝性神経変性疾患の遺伝子操作:神話か現実か? Br. J.Ophthalmol. 100:1322. 土井:10.1136/ビョフタルモル-2015-308329
PubMed Abstract|CrossRef全文|Google Scholar
Zhao,Z.,Zhang,X.,Zhao,C.,Choi,J.,Shi,J.,Song,K.、ら。 (2010). ホスホリパーゼA(2)を介したグループによる膵β細胞の保護-ミトコンドリア膜過酸化の修復。 内分泌学151、3038-3048。 土井:10.1210/en.2010-0016
PubMed Abstract|CrossRef Full Text|Google Scholar