- Introduction
- Pathologie moléculaire
- Diagnostic moléculaire et Autonomisation des patients atteints de maladies rares
- Thérapies possibles
- Thérapie de remplacement enzymatique (TRE)
- Thérapie génique / Remplacement génique
- Modification du gène/de la base
- Conclusion
- Contributions des auteurs
- Déclaration de conflit d’intérêts
Introduction
La dystrophie neuroaxonale infantile (DNA) est une maladie neurodégénérative rare autosomique récessive de fréquence inconnue. L’apparition des symptômes survient généralement entre 6 mois et 3 ans. Avant cette période, les nourrissons se développent normalement. Le premier symptôme de l’INAD peut être le ralentissement de l’atteinte des jalons de développement normaux ou la régression des jalons de développement (Ramanadham et al., 2015). L’hypotonie du tronc, le strabisme et le nystagmus sont les premiers symptômes de la maladie (Gregory et al., 2017). La progression de la maladie est rapide et à mesure qu’elle progresse, davantage de compétences acquises sont perdues. Les muscles deviennent rapidement hypotoniques et deviennent plus tard spastiques (Levi et Finazzi, 2014). Finalement, tout contrôle musculaire volontaire est perdu. La faiblesse musculaire peut également entraîner des difficultés d’alimentation et de respiration. En plus du nystagmus, certains enfants souffrent d’une perte de vision. Les fonctions cognitives sont progressivement perdues et la démence se développe. La durée de vie est généralement de 5 à 10 ans (Gregory et al., 1993; Jardim et coll., 2004; Macauley et Sands, 2009).
Pathologie moléculaire
La dystrophie neuroaxonale infantile appartient à une famille de troubles neurodégénératifs qui comprend la dystrophie neuroaxonale atypique à début tardif (ANAD) et le complexe de parkinsonisme de dystonie (DPC). La plupart des cas d’INAD sont associés à des mutations homozygotes ou hétérozygotes composées du gène PLA2G6 qui affectent l’activité catalytique de son produit protéique (Engel et al., 2010). Le gène PLA2G6 code un groupe via une protéine phospholipase A2 indépendante du calcium (PLA2G6 ou iPLA2ß, ∼85/88 kDa) avec une lipase et sept domaines contenant des répétitions d’ankyrine (Tang et al., 1997). Le PLA2G6 hydrolyse la chaîne acyle sn-2 des phospholipides, générant des acides gras libres et des lysophospholipides.
Les phospholipides de la membrane interne des mitochondries sont riches en acides gras insaturés en position sn-2, en particulier en cardiolipine (Seleznev et al., 2006). Ces acides gras insaturés sont particulièrement vulnérables aux espèces réactives abondantes de l’oxygène produites par les mitochondries (Murphy, 2009), ce qui entraîne des phospholipides peroxydés dans la membrane interne des mitochondries. Le PLA2G6 se localise dans les mitochondries (Williams et Gottlieb, 2002; Liou et al., 2005) en accord avec une demande accrue d’hydrolyse des acides gras peroxydés en position sn-2 des phospholipides conduisant à des phospholipides remodelés (Balsinde et al., 1995; Zhao et coll., 2010). Lorsque le PLA2G6 est défectueux, l’intégrité de la membrane interne des mitochondries est endommagée. PLA2G6 se localise également à l’axone (Ong et al., 2005; Seleznev et coll., 2006) indiquant une demande localisée accrue pour le remodelage des phospholipides là aussi. La manifestation d’une telle accumulation dans le cerveau est unique à des zones clés du cerveau, telles que les ganglions de la base, ce qui a donné divers noms à la même pathogenèse moléculaire sous-jacente impliquant PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
L’analyse ultrastructurale des neurones de souris PLA2G6 knockout est compatible avec cette pathologie moléculaire. Des mitochondries avec des cristées ramifiées et tubulaires, des mitochondries avec des cristées dégénérées, des axones avec effondrement du cytosquelette et une perte partielle de la membrane aux terminaisons des axones ont été observées (Beck et al., 2011). Au niveau microscopique, ces caractéristiques apparaissent sous forme de gonflements axonaux et de corps sphéroïdes dans les terminaux pré-synaptiques (Figure 1) du système nerveux central ou périphérique.
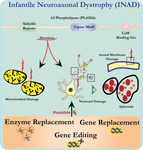
Figure 1. La dystrophie neuroaxonale infantile (DAIN) est une maladie neurodégénérative liée à des mutations du gène PLA2G6. Diverses mutations de la PLA2G6 conduisent à une phospholipase A2 dysfonctionnelle qui entraîne des défauts de la membrane mitochondriale et axonale. Ces défauts provoquent des lésions neuronales visualisées sous forme de gonflements axonaux et d’accumulation de sphéroïdes pré-synaptiques. Le remplacement enzymatique pour restaurer les fonctions, le remplacement génique ou l’édition pour corriger le PLA2G6 défectueux sont des stratégies thérapeutiques proposées.
Diagnostic moléculaire et Autonomisation des patients atteints de maladies rares
Outre des caractéristiques cliniques, électrophysiologiques et d’imagerie spécifiques, avant la disponibilité du séquençage de nouvelle génération, des biopsies cutanées montrant des gonflements axonaux et des corps sphéroïdes dans des terminaux pré-synaptiques du système nerveux central ou périphérique étaient les critères diagnostiques de confirmation de l’INAD (Gregory et al., 2017; Iodice et coll., 2017). Souvent, plusieurs biopsies étaient nécessaires pour confirmer le diagnostic. Les familles ont généralement attendu de nombreuses années pour obtenir un diagnostic. Avec la diminution du coût du séquençage des gènes et du génome, la disponibilité de tests ciblés par panel de gènes dans les laboratoires de diagnostic pour les maladies neurologiques non diagnostiquées et la sensibilisation croissante des médecins à la disponibilité des diagnostics génétiques, les familles reçoivent un diagnostic plus rapidement; parfois dans l’année suivant l’apparition du premier symptôme. Les enfants de ces familles sont encore jeunes et les familles sont motivées à s’associer à des scientifiques pour trouver un traitement pour la maladie de leurs enfants. Afin de financer le processus de recherche, un groupe de parents de patients INAD a formé la Fondation INADcure. La fondation a recueilli des fonds substantiels pour la recherche et s’associe à l’Institut de génomique rare pour les guider dans l’octroi de subventions de recherche. Fait intéressant, une analyse génétique de 2016 de 22 familles indiennes avec INAD, ANAD et DPC a révélé que 10/22 familles (45,45%) manquaient de mutations dans la région codante du gène PLA2G6 (Kapoor et al., 2016). L’échec de l’identification de mutations délétères dans la région codante du PLA2G6 met en évidence que les futurs efforts de diagnostic moléculaire nécessiteraient un séquençage génétique complet pour identifier les mutations dans les régions introniques et régulatrices du gène PLA2G6 chez les patients atteints d’INAD. De plus, dans les cas où aucune mutation associée au PLA2G6 n’est observée, la cause de la maladie pourrait être due à une mutation de gènes autres que le PLA2G, qui doit être explorée.
Thérapies possibles
La plupart des thérapies envisagées pour une maladie rare comme l’INAD impliquant une enzyme défectueuse sont le remplacement enzymatique, le remplacement génique ou la correction génique. Lorsqu’un déficit enzymatique est causé par un défaut génétique récessif, on suppose que le remplacement ou la supplémentation enzymatique peut corriger le problème (Smith et al., 2012; Yu-Wai-Man, 2016). Cependant, des expériences visant à prouver que des thérapies fournissant le gène ou l’enzyme correct sauveront le phénotype INAD doivent encore être effectuées et testées.
Thérapie de remplacement enzymatique (TRE)
Étant donné que le cerveau est l’organe principal affecté par l’INAD, la thérapie de remplacement enzymatique de l’INAD nécessiterait très probablement une perfusion de l’enzyme dans le cerveau. En 2017, Biomarin a reçu l’approbation de la FDA pour la tripeptidyl peptidase 1 (cerliponase alfa) en tant que traitement de la cause sous-jacente de la maladie de Batten, la carence en tripeptidyl peptidase (TPP1) une enzyme lysosomale (U.S. Food Drug Administration, 2017). Tripeptidyl peptidase 1 (cérliponase alfa; 59 kDa) est la première ERT à être directement administrée dans le liquide céphalo-rachidien (LCR) du cerveau. D’autres médicaments ERT qui peuvent être administrés dans le LCR font l’objet d’essais cliniques (Jardim et al., 2004; Macauley et Sands, 2009).
L’efficacité de la tripeptidyl peptidase 1 (cérliponase alfa) sur la capacité de marche des patients atteints de la maladie de latte démontre qu’un traitement de remplacement enzymatique pour les maladies affectant le cerveau est théoriquement possible. Cibler l’enzyme de remplacement dans les mitochondries pour l’INAD sera plus difficile que le ciblage lysosomal qui avait déjà été réussi par l’ERT IV depuis longtemps comme dans le cas de la maladie de Gaucher. Par conséquent, de nombreux problèmes biologiques spécifiques à l’INAD doivent encore être résolus:
Un autre problème est que la thérapie de remplacement enzymatique nécessite la mise en place initiale d’un cathéter intracérébroventriculaire et des perfusions fréquentes. La mise en place du cathéter nécessite une anesthésie et les perfusions peuvent nécessiter une anesthésie en fonction de la coopération des patients. Répondre aux préoccupations des neurologues pédiatriques et des parents d’enfants atteints d’INAD en ce qui concerne l’utilisation de l’anesthésie chez les patients atteints d’INAD avec des informations appropriées nécessitera des efforts supplémentaires.
Thérapie génique / Remplacement génique
Le gène PLA2G humain est d’environ 70 kb avec 17 exons et 2 exons alternatifs. Cependant, la transcription codante pour les protéines la plus longue n’est que de 3,3 ko et la séquence codante pour les protéines est d’un peu plus de 2,4 ko, ce qui peut facilement être emballé dans une cargaison de vecteurs viraux. Nous explorerons d’abord un autre trouble du stockage lysosomal dans le SNC qui possède des données précliniques à comparer ensuite à l’INAD. Le manque de données précliniques de l’ERT dans l’INAD est une lacune importante dans les connaissances sur le terrain, c’est pourquoi il est crucial que nous observions d’autres modèles précliniques de troubles du SNC qui font l’objet d’études de thérapie génique. Auparavant, une étude préclinique chez la souris portait sur la thérapie génique du trouble du stockage lysosomal des mucopolysaccharidoses de type IIIA (MPS-IIIA) en conditionnant la version correcte du gène de la sulfamidase à l’intérieur d’un vecteur viral (Sorrentino et al., 2013). L’étude a profité de la compréhension croissante de la barrière hémato-encéphalique (BBB) qui régule activement le transport de grosses molécules du sang vers le SNC par un processus appelé transcytose (Pardidge, 2005b; Sorrentino et Fraldi, 2016).
La transcytose implique une endocytose de ligands du côté luminal, médiée par des récepteurs spécifiques au ligand (par exemple, récepteur de l’insuline, récepteur de la transferrine et récepteur des lipoprotéines de basse densité, etc.,) enrichi sur l’endothélium capillaire (Pardridge, 2005a). Ceci est suivi d’un mouvement de la cargaison endocytée à travers le cytoplasme de l’endothélium et enfin d’une exocytose du côté abluminal (cerveau), livrant ainsi efficacement la cargaison à travers le BBB (Pardridge, 2002). L’étude préclinique MPS-IIIA a fabriqué une protéine sulfamidase chimérique contenant un domaine de liaison BBB de l’apolipoprotéine B pour faciliter l’absorption par l’endothélium et également un peptide signal de l’iduronate-2-sulfatase pour faciliter une exocytose efficace vers le côté abluminal de la BBB (Sorrentino et al., 2013). Une cargaison de vecteur viral codant pour une sulfamidase chimérique a ensuite été chargée sur un virus adéno-associé (AAV) sérotype 2/8 ciblant le foie (Sorrentino et Fraldi, 2016). Ainsi, le foie a servi d’usine interne fournissant un approvisionnement constant en chimérique-sulfamidase, ce qui a entraîné une augmentation de 10 à 15% de l’activité de la sulfamidase cérébrale, même 7 mois après la thérapie génique hépatique. Cette augmentation des niveaux d’activité de la sulfamidase cérébrale a conduit à une amélioration quantifiable des résultats de la pathologie cérébrale et du comportement dans le modèle murin de MPS-IIIA (Sorrentino et al., 2013).
Si une étude similaire est menée pour l’INAD, elle répondrait à plusieurs questions critiques sur le remplacement enzymatique du traitement par l’INAD. Alternativement, l’administration intra-vasculaire ou intra-LCR de produits de thérapie génique à base d’AAV9 peut cibler directement le SNC (Bey et al., 2017; Roca et coll., 2017). Cependant, les multiples mutations que les patients INAD ont sur leur gène PLAG26 posent des défis uniques à la thérapie génique. Bien que la taille du gène PLA2G6 de 2-3 kb ne devrait pas poser de problème pour son insertion dans un vecteur viral, cependant, les complications réglementaires liées à la correction du PLA2G6 sont difficiles à prévoir. Les complications régulatrices deviennent incontrôlables surtout si la thérapie génique ne peut pas être localisée correctement dans les tissus cibles. La thérapie de remplacement enzymatique peut également poser problème si certains produits mutants s’avèrent être négatifs dominants pour le PLAG26 de type sauvage.
La plupart des défis des thérapies possibles pour l’INAD viennent du fait qu’il s’agit d’une maladie ultra-rare. Cependant, le fait que l’INAD ait une étiologie génétique connue offre des pistes de thérapies possibles. De plus, toute thérapie réussie pour l’INAD recevra le statut de médicament orphelin et toute la protection qu’elle obtient car l’INAD est une maladie rare. Ainsi, malgré tous les défis susmentionnés, le statut de médicament orphelin constitue une forte incitation pour les chercheurs en maladies rares et l’industrie de la biotechnologie, sans parler lorsque la cause génétique est déjà connue.
Modification du gène/de la base
En 2017, au moins 277 mutations faux sens ont été observées dans le gène humain PLA2G6 (Lek et al., 2016). Seule une faible proportion des mutations PLA2G6 comprennent des décalages de cadre, des indels, des mutations non-sens et des mutations dans les sites d’épissure (Morgan et al., 2006). Ainsi, corriger le gène dans la population de cellules cibles ou corriger les bases d’ADN mutées est une piste thérapeutique intéressante. Au fil des ans, plusieurs outils ont été développés pour l’édition de gènes, et l’édition du génome basée sur CRISPR/Cas9 est saluée comme une technologie révolutionnaire avec un essai clinique prévu en 2018. Ces technologies utilisent une protéine de liaison à l’ADN qui peut également cliver le brin de manière spécifiée pour laisser de la place à l’insertion d’une nouvelle séquence d’ADN ou à la correction de la base d’ADN délétère (LaFountaine et al., 2015). La technologie CRISPR/Cas9 a déjà donné des résultats thérapeutiques étonnants dans plusieurs modèles précliniques de maladies (par exemple, la dystrophie musculaire de Duchene, les maladies métaboliques du foie, etc.,) (Dai et coll., 2016). De plus, une compréhension croissante de la variation humaine pose de nouveaux défis à la thérapie génique et pousse l’innovation vers une véritable personnalisation de l’édition génique (Lessard et al., 2017; Scott et Zhang, 2017). Les nouvelles technologies telles que vSLENDR, un virus AAV, et la technologie médiée par CRISPR/Cas9 pour remplacer les gènes défectueux dans les neurones et d’autres cellules du système nerveux poussent les technologies d’édition de gènes à de nouvelles frontières (Nishiyama et al., 2017).
Conclusion
La dystrophie neuroaxonale infantile est une maladie neurodégénérative sévère avec une certaine morbidité et mortalité. Cette maladie rare offre une opportunité passionnante de revalider les modes de thérapie disponibles de la prochaine génération et d’en générer de nouveaux. La nouvelle congruence sur les critères de diagnostic clinique de l’INAD devrait donner une impulsion à l’amélioration du diagnostic moléculaire. Ces progrès devraient nous conduire à développer des thérapies abordables qui permettraient d’améliorer quantifiablement la qualité de vie des patients atteints d’INAD et de retarder ou d’améliorer la progression de la maladie. Nous avons mis en évidence quelques réussites dans la maladie de Batten et les mucopolysaccharidoses qui nous inspirent à poser les bonnes questions pour faire de l’INAD-thérapie une réalité. De plus, des approches virales et non virales croissantes pour l’édition de gènes à base de CRISPR / Cas9 devraient également ouvrir de nouvelles voies thérapeutiques pour l’INAD.
Contributions des auteurs
PB et FA ont préparé un avant-projet. SR et AC ont édité et ajouté d’autres sections au manuscrit. DF, AP et LP ont édité le manuscrit.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., et Dennis, E. A. (1995). L’inhibition de la phospholipase A2 indépendante du calcium empêche l’incorporation d’acide arachidonique et le remodelage des phospholipides dans les macrophages P388D1. Proc. Natl. Acad. Sci. États-Unis 92, 8527-8531. doi: 10.1073/pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Ciblage efficace du SNC chez des souris adultes par perfusion intrathécale d’AAV9-GFP monocaténaire pour la thérapie génique des troubles neurologiques. Gene Ther. 24:325. doi: 10.1038/gt.2017.18
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 pour la thérapie génique in vivo: promesse et obstacles. Mol. Là. Acides nucléiques 5: e349. doi: 10.1038/ mtna.2016.58
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Engel, L. A., Jing, Z., O’Brien, D. E., Sun, M., et Kotzbauer, P.T. (2010). La fonction catalytique du PLA2G6 est altérée par des mutations associées à la dystrophie neuroaxonale infantile, mais pas à la dystonie-parkinsonisme. PLoS Un 5: e12897. doi: 10.1371 / journal.pone.0012897
Résumé PubMed / CrossRef Texte intégral
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., et Hayflick, S. J. (1993). dans la neurodégénérescence associée à PLA2G6, eds M. P. Adam, H. H. Ardinger et R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Implication du SNC dans la maladie de Fabry: études cliniques et d’imagerie avant et après 12 mois de thérapie de remplacement enzymatique. J. Hériter. Métab. Dis. 27, 229–240. doi: 10.1023/B: BOLI.0000028794.04349.91
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Rather, M. I., Bhat, V., Gopinath, S., et al. (2016). Analyse génétique du PLA2G6 dans 22 familles indiennes atteintes de dystrophie neuroaxonale infantile, de dystrophie neuroaxonale atypique à début tardif et de complexe de parkinsonisme de dystonie. PLoS One 11: e0155605. doi: 10.1371 / journal.pone.0155605
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit de l’un des principaux ouvrages de l’auteur. Livraison et applications thérapeutiques des technologies d’édition de gènes ZFNs. TALENs et CRISPR/Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016/ j. ijpharm.2015.08.029
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291. doi: 10.1038 / nature19057
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). La variation génétique humaine modifie la spécificité de ciblage sur et hors ciblage de CRISPR-Cas9 au niveau des loci impliqués sur le plan thérapeutique. Proc. Natl. Acad. Sci. États–Unis 14, E11257-E11266. doi: 10.1073/pnas.1714640114
Résumé PubMed / Texte intégral Croisé / Google Scholar
Levi, S., et Finazzi, D. (2014). Neurodégénérescence avec accumulation de fer dans le cerveau: mise à jour sur les mécanismes pathogènes. Devant. Pharmacol. 5:99. doi: 10.3389/ fphar.2014.00099
CrossRef Texte intégral / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Localisation mitochondriale de la cyclooxygénase-2 et de la phospholipase A2 indépendante du calcium dans les cellules cancéreuses humaines: implication dans la résistance à l’apoptose. Exp. Cellule 306, 75-84. doi: 10.1016/j.yexcr.2005.01.011
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Macauley, S.L., et Sands, M.S. (2009). Thérapie de remplacement enzymatique dirigée par le SNC prometteuse pour les maladies de stockage lysosomales. Exp. Neurol. 218, 5–8. doi: 10.1016/ j. expneurol.2009.03.040
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Mehnaaz, L. (2016). Neurodégénérescence avec accumulation de fer dans le cerveau (NBIA) anciennement maladie de Hallervorden-Spatz. J. Assoc. Phys. Inde 64:132.
Google Scholar
Il s’agit de l’un des principaux facteurs de risque de la maladie, notamment le cancer du sein, le cancer du sein, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson, la maladie de Parkinson et la maladie de Parkinson. (2006). La PLA2G6, codant pour une phospholipase A2, est mutée dans les troubles neurodégénératifs avec une teneur élevée en fer cérébral. NAT. Genet. 38, 752–754. doi: 10.1038/ ng1826
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Murphy, député (2009). Comment les mitochondries produisent des espèces réactives de l’oxygène. Biochem. J. 417, 1-13. doi: 10.1042/BJ20081386
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C. et Faria, A. (2016). Neurodégénérescence avec accumulation de fer dans le cerveau: rapport de cas. Dement. Neuropsychol. 10, 160–164. doi: 10.1590/S1980-5764-2016DN1002014
Résumé PubMed / Texte intégral croisé / Google Scholar
Il s’agit de la première édition de la série. Édition du génome à médiation virale via une réparation dirigée par homologie dans les cellules mitotiques et postmitotiques du cerveau des mammifères. Neurone 96, 755.e5-768.e5. doi: 10.1016/j. neurone.2017.10.004
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F. et Farooqui, AA (2005). Distribution de la phospholipase A2 indépendante du calcium (iPLA2) dans le cerveau de singe. J. Neurocytol. 34, 447–458. doi: 10.1007/s11068-006-8730-4
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Pardidge, W. M. (2002). Ciblage de médicaments et de gènes vers le cerveau avec des chevaux de Troie moléculaires. NAT. Le révérend Drug Discov. 1, 131–139. doi: 10.1038 /nrd725
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Pardidge, W. M. (2005a). Biologie moléculaire de la barrière hémato-encéphalique. Mol. Biotechnol. 30, 57–70. doi: 10.1385/MB:30:1:057
CrossRef Texte intégral / Google Scholar
Pardidge, W. M. (2005b). La barrière hémato-encéphalique: goulot d’étranglement dans le développement de médicaments cérébraux. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Phospholipases A2 indépendantes du calcium et leurs rôles dans les processus biologiques et les maladies. J. Lipid Res. 56, 1643-1668. doi: 10.1194/jlr.R058701
Résumé PubMed / Texte intégral Croisé / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Correction de la maladie par thérapie génique médiée par l’AAV dans un nouveau modèle murin de mucopolysaccharidose de type IIID. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg/ddx058
Résumé PubMed / Texte intégral Croisé / Google Scholar
Scott, D.A., et Zhang, F. (2017). Implications of human genetic variation in CRISPR-based therapeutic genome editing. NAT. Med. 23:1095. doi: 10.1038/nm.4377
Résumé PubMed / CrossRef Texte intégral / Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Chanson, K., et Ma, Z. A. (2006). La phospholipase A2, indépendante du calcium, localise et protège les mitochondries lors de l’induction apoptotique par la staurosporine. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
Résumé PubMed / Texte intégral Croisé / Google Scholar
Il s’agit de la première édition de la série. Thérapie par supplémentation génétique pour les formes récessives de dystrophies rétiniennes héréditaires. Gene Ther. 19, 154–161. doi: 10.1038/gt.2011.161
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Sorrentino, N. C., D’Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Une sulfamidase hautement sécrétée conçue pour traverser la barrière hémato-encéphalique corrige les lésions cérébrales de souris atteintes de mucopolysaccharidoses de type IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002/emmm.201202083
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Sorrentino, N. C., et Fraldi, A. (2016). Ciblage du cerveau dans MPS-IIIA. Pédiatre. Endocrinol. Rév.13 (Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., et Jones, S.s. (1997). Une nouvelle phospholipase A2 cytosolique indépendante du calcium contient huit motifs d’ankyrine. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Food and Drug Administration des États-Unis (2017). La FDA approuve le premier traitement pour une forme de maladie des lattes. Disponible à: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., et Gottlieb, R.A. (2002). L’inhibition de la phospholipase A2 indépendante du calcium mitochondrial (iPLA2) atténue la perte de phospholipides mitochondriaux et est cardioprotectrice. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042/bj3620023
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Yu-Wai-Man, P. (2016). Manipulation génétique pour les maladies neurodégénératives héréditaires: mythe ou réalité? Fr. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophtalmol-2015-308329
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et coll. (2010). Protection des cellules bêta pancréatiques par groupe VIA la réparation médiée par la phospholipase A (2) de la peroxydation de la membrane mitochondriale. Endocrinologie 151, 3038-3048. doi: 10.1210 / fr.2010-0016
Résumé PubMed / Texte intégral CrossRef / Google Scholar