Introduction
infantiili neuroaksonaalinen dystrofia (Inad) on autosomaalisesti resessiivisesti resessiivinen harvinainen neurodegeneratiivinen sairaus, jota ei tunneta. Oireiden alkaminen tapahtuu yleensä 6 kuukauden ja 3 vuoden iässä. Sitä ennen pikkulapset kehittyvät normaalisti. Inad: n ensimmäinen oire voi olla normaalien kehityshäiriöiden virstanpylväiden saavuttamisen hidastuminen tai taantuminen kehityshäiriöiden virstanpylväissä (Ramanadham et al., 2015). Rungon hypotonia, karsastus ja nystagmus ovat taudin varhaisia oireita (Gregory et al., 2017). Taudin eteneminen on nopeaa ja sen edetessä menetetään lisää hankittuja taitoja. Lihakset muuttuvat pian hypotonisiksi ja myöhemmin spastisiksi (Levi ja Finazzi, 2014). Lopulta kaikki vapaaehtoinen lihashallinta menetetään. Lihasheikkous voi johtaa myös ruokinnan ja hengityksen vaikeutumiseen. Nystagmuksen lisäksi jotkut lapset kokevat näön heikkenemistä. Kognitiiviset toiminnot häviävät vähitellen ja dementia kehittyy. Elinikä on yleensä 5-10 vuotta (Gregory et al., 1993; Jardim ym., 2004; Macauley and Sands, 2009).
Molekyylipatologia
infantiili Neuroaksonaalinen dystrofia kuuluu neurodegeneratiivisten häiriöiden ryhmään, johon kuuluvat epätyypilliset myöhäisen neuroaksonaaliset dystrofiat (Anad) ja dystonia Parkinsonismikompleksi (DPC). Useimmat inad-tapaukset liittyvät HOMOTSYGOOTTISIIN tai yhdisteen heterotsygoottisiin mutaatioihin PLA2G6-geenissä, jotka vaikuttavat sen proteiinituotteen katalyyttiseen aktiivisuuteen (Engel ym., 2010). PLA2G6-geeni koodaa ryhmän kalsiumriippumattoman fosfolipaasi A2-proteiinin (PLA2G6 tai iPLA2ß, ∼85/88 kDa) kautta lipaasin ja seitsemän ankyriinin toistoa sisältävän domeenin (Tang et al., 1997). PLA2G6 hydrolysoi fosfolipidien Sn-2-asyyliketjun tuottaen vapaita rasvahappoja ja lysofosfolipidejä.
mitokondrioiden sisäkalvon Fosfolipideissä on runsaasti Sn-2-asemassa olevia tyydyttymättömiä rasvahappoja, erityisesti kardiolipiiniä (Seleznev et al., 2006). Nämä tyydyttymättömät rasvahapot ovat erityisen alttiita mitokondrioiden tuottamille runsaille reaktiivisille happilajeille (Murphy, 2009), jotka johtavat mitokondrioiden sisäkalvon peroksidoituneisiin fosfolipideihin. PLA2G6 paikallistaa mitokondrioihin (Williams and Gottlieb, 2002; Liou et al., 2005), joka on sopusoinnussa fosfolipidien Sn-2-asemassa olevien peroksidoitujen rasvahappojen hydrolyysin lisääntyneen kysynnän kanssa, mikä johtaa uudelleenmuotoutuneisiin fosfolipideihin (Balsinde et al., 1995; Zhao et al., 2010). Kun PLA2G6 on viallinen, mitokondrion sisäkalvon eheys vaurioituu. PLA2G6 paikallistaa myös aksonille (Ong et al., 2005; Seleznev et al., 2006) osoittaa, että fosfolipidien remodelointi on lisääntynyt paikallisesti sielläkin. Tällaisen kertymisen ilmentyminen aivoissa on ainutlaatuinen keskeisillä aivoalueilla, kuten tyvitumakkeessa, joka johti eri nimiin samalle taustalla olevalle molekyylipatogeneesille, johon liittyy PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
ULTRARAKENNE analyysi neuronien PLA2G6 knockout hiirillä on yhdenmukainen tämän molekyylipatologian kanssa. On havaittu mitokondrioita, joissa on haarautuvia ja putkimaisia cristaeita, mitokondrioita, joissa on degeneroituneita cristaeita, aksoneita, joissa on sytoskeletoni romahtaa, ja osittainen kalvon menetys aksonipäätteissä (Beck et al., 2011). Mikroskooppisella tasolla nämä piirteet ilmenevät aksonaalisina turvotuksina ja pallomaisina kehoina keskus-tai ääreishermoston pre-synaptisissa terminaaleissa (Kuva 1).
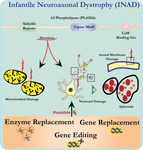
Kuva 1. Infantiili neuroaksonaalinen dystrofia (INAD) on neurodegeneratiivinen häiriö, joka liittyy PLA2G6-geenin mutaatioihin. PLA2G6: n erilaiset mutaatiot johtavat toimintahäiriöiseen A2-fosfolipaasiin, joka johtaa mitokondrion ja aksonaalisen kalvon virheisiin. Nämä viat aiheuttavat neuronaalisia vaurioita, jotka näkyvät aksonaalisina turvotuksina ja esisynaptisten sferoidien kertymisenä. Entsyymin korvaaminen toimintojen palauttamiseksi, geenien korvaaminen tai muokkaaminen viallisen PLA2G6: n korjaamiseksi ehdotetaan terapeuttisia strategioita.
Molekyylidiagnostiikka ja harvinaisten sairauksien potilaiden voimaantuminen
lukuun ottamatta erityisiä kliinisiä, elektrofysiologisia ja kuvantamisominaisuuksia, ennen seuraavan sukupolven sekvensoinnin saatavuutta ihon koepalat, joissa näkyi aksonaalisia turvotuksia ja pallomaisia kappaleita esisynaptisissa terminaaleissa keskus-tai ääreishermostossa, olivat diagnostiset kriteerit inad: n vahvistamiselle (Gregory et al., 2017; Jodice et al., 2017). Diagnoosin varmistamiseksi tarvittiin usein useita koepaloja. Perheet odottivat yleensä diagnoosia monta vuotta. Kun geeni-ja genomisekvensoinnin kustannukset pienenevät, diagnosoimattomien neurologisten sairauksien diagnostisten laboratorioiden kanssa tehtävät kohdennetut geenipaneelitestit ja lääkäreiden tietoisuus geneettisen diagnostiikan saatavuudesta lisääntyvät, perheet saavat diagnoosin nopeammin; joskus vuoden kuluessa ensimmäisestä oireesta. Näiden perheiden lapset ovat vielä nuoria ja perheet ovat motivoituneita yhteistyöhön tutkijoiden kanssa löytääkseen hoitoa lastensa sairauteen. Tutkimusprosessin rahoittamiseksi joukko inad-potilaiden vanhempia perusti Inad-säätiön. Säätiö on kerännyt huomattavia varoja tutkimukseen ja toimii yhteistyössä Rare Genomics Instituten kanssa opastaakseen heitä tutkimusapurahojen myöntämisessä. Mielenkiintoista, 2016 geneettinen analyysi 22 Intian perheet INAD, ANAD, ja DPC todettiin, että 10/22 perheet (45.45%) puuttui mutaatioita PLA2G6 geenin koodaus alueella (Kapoor et al., 2016). Jos PLA2G6: n koodausalueella ei tunnisteta haitallisia mutaatioita, korostuu, että tulevaisuudessa molekyylidiagnostiikkatoimet vaatisivat koko geenisekvenssiä mutaatioiden tunnistamiseksi PLA2G6-geenin intronisilla ja säätelevillä alueilla INAD-tautia sairastavilla potilailla. Lisäksi inad-tapaukset, joissa PLA2G6: een liittyviä mutaatioita ei havaita, viittaavat siihen, että taudin syy voi johtua mutaatiosta muissa geeneissä kuin PLA2G: ssä, mikä on tutkittava.
mahdolliset hoidot
useimmat niistä hoidoista, joita pidetään harvinaiseen sairauteen, kuten inad: iin, jossa on viallinen entsyymi, ovat entsyymikorjaus, geenikorjaus tai geenikorjaus. Kun entsyymin puutos johtuu resessiivisestä geenivirheestä, oletetaan, että entsyymin korvaaminen tai täydentäminen voi korjata ongelman (Smith et al., 2012; Yu-Wai-Man, 2016). On kuitenkin vielä tehtävä ja testattava kokeita, joilla todistetaan, että oikean geenin tai entsyymin tuottavat hoidot pelastavat inad-fenotyypin.
entsyymikorvaushoito (ERT)
koska inad vaikuttaa ensisijaisesti aivoihin, inad: n entsyymikorvaushoito vaatisi mitä todennäköisimmin entsyymin infuusion aivoihin. Vuonna 2017 Biomariini sai FDA: n hyväksynnän tripeptidyylipeptidaasi 1: lle (cerliponaasialfa) hoitona Battenin taudin perussyylle, tripeptidyylipeptidaasin (TPP1) lysosomaalisen entsyymin puutokselle (U. S. Food Drug Administration, 2017). Tripeptidyylipeptidaasi 1 (serliponaasialfa; ∼59 kDa) on ensimmäinen ERT, joka annetaan suoraan aivo-selkäydinnesteeseen (CSF). Muut ERT-lääkkeet, joita voidaan antaa aivo-selkäydinnesteeseen, ovat kliinisissä tutkimuksissa (Jardim et al., 2004; Macauley and Sands, 2009).
tripeptidyylipeptidaasi 1: n (serliponaasialfa) teho Battenin tautia sairastavien potilaiden kävelykykyyn osoittaa, että aivoihin vaikuttavien sairauksien entsyymikorvaushoito on teoriassa mahdollista. Korvaavan entsyymin kohdentaminen inad: n mitokondrioihin on haastavampaa kuin lysosomaalinen kohdistaminen, joka oli jo pitkään onnistunut IV ERT: llä, kuten Gaucherin taudin tapauksessa. Siksi on vielä monia inad: lle ominaisia biologisia kysymyksiä, jotka on ratkaistava:
• tarvitseeko inad: n hoito mekanismeja, jotka voivat mahdollisesti kuljettaa PLA2G6: n soluun?
• onko ääreishermoston kohdistamiseen erityistä tarvetta?
lisäksi on kyse siitä, että entsyymikorvaushoito edellyttää intrakerebroventrikulaarisen katetrin alustavaa sijoittamista ja toistuvia infuusioita. Katetrin sijoittaminen vaatii puudutusta ja infuusiot voivat vaatia puudutusta riippuen potilaiden yhteistyöstä. Lastenneurologien ja INAD-tautia sairastavien lasten vanhempien huolenaiheiden käsitteleminen nukutuksen käytöstä INAD-potilailla asianmukaisten tietojen saamiseksi vaatii lisätoimia.
geeniterapia / Geeninkorvaus
ihmisen PLA2G-geeni on noin 70 kb, jossa on 17 eksonia ja 2 vaihtoehtoista eksonia. Pisin proteiinia koodaava transkriptio on kuitenkin vain 3,3 kb ja proteiinin koodaussekvenssi hieman yli 2,4 kb, joka voidaan helposti pakata virusvektorilastiin. Tutkimme ensin toista LYSOSOMAALISTA varastointihäiriötä keskushermostossa, jolla on prekliinisiä tietoja, joita sitten verrataan INAD: hen. ERT: n prekliinisten tietojen puute INAD: ssä on merkittävä tietämyskuilu alalla, minkä vuoksi on erittäin tärkeää, että tarkkailemme muita KESKUSHERMOSTOHÄIRIÖIDEN prekliinisiä malleja, joita tutkitaan geeniterapiassa. Aiemmin prekliinisessä hiirillä tehdyssä tutkimuksessa käsiteltiin lysosomaalisen säilytyshäiriön mukopolysakkaridoosien tyypin IIIA (MPS-IIIA) geeniterapiaa pakkaamalla sulfamidaasigeenin oikea versio virusvektorin sisään (Sorrentino et al., 2013). Tutkimuksessa hyödynnettiin kasvavaa ymmärrystä veri – aivoesteestä (BBB), joka säätelee aktiivisesti suurten molekyylien kuljetusta verestä keskushermostoon transcytosis-prosessilla (Pardridge, 2005b; Sorrentino and Fraldi, 2016).
Transkytoosiin liittyy ligandispesifisten reseptorien (esim.insuliinireseptori, transferriinireseptori ja low-density-lipoproteiinireseptori) välittämien luminaalipuolen ligandien endosytoosi.,) rikastettu kapillaari endoteeli (Pardridge, 2005a). Tätä seuraa endoteelisytoplasman siirtyminen endoteelisytoplasman läpi ja lopulta eksosytoosi abluminaalipuolella (aivot), jolloin lasti siirtyy tehokkaasti BBB: n poikki (Pardridge, 2002). MPS-IIIA prekliininen tutkimus valmisti kimeeristä sulfamidaasiproteiinia, joka sisälsi apolipoproteiini B: stä BBB: tä sitovan domeenin endoteelin sisäänoton helpottamiseksi ja myös signaalipeptidin iduronaatti-2-sulfataasista tehokkaan eksosytoosin edistämiseksi BBB: n abluminaalista puolta kohti (Sorrentino et al., 2013). Kimeeristä sulfamidaasia koodaava virusvektorilasti lastattiin sitten adenoon liittyvään serotyyppiin 2/8 (AAV), joka kohdistuu maksaan (Sorrentino and Fraldi, 2016). Näin maksa toimi sisäisenä tehtaana, joka tarjosi jatkuvasti kimeerisulfamidaasia, mikä johti 10-15%: n kasvuun aivojen sulfamidaasiaktiivisuudessa jopa 7 kuukautta maksan geenihoidon jälkeen. Tämä aivojen sulfamidaasiaktiivisuuden lisääntyminen johti aivojen patologian ja käyttäytymisen tulosten kvantitoitavaan paranemiseen hiirimallissa MPS-IIIA (Sorrentino et al., 2013).
jos vastaava tutkimus tehdään INAD: lle, se vastaisi useisiin kriittisiin kysymyksiin inad-hoidon entsyymikorvauksesta. Vaihtoehtoisesti aiv9-pohjaisten geeniterapiatuotteiden suonensisäinen tai aivo-selkäydinnesteen sisäinen anto voi kohdistua suoraan keskushermostoon (Bey et al., 2017; Roca et al., 2017). Kuitenkin, useita mutaatioita, jotka INAD potilailla on heidän PLAG26 geeni tarjoaa ainutlaatuisia haasteita geeniterapian. Vaikka 2-3 kb: n PLA2G6-geenin koon ei pitäisi aiheuttaa ongelmaa sen asettamisessa virusvektoriin, kuitenkin pla2g6: n korjaamisesta aiheutuvia sääntelykomplikaatioita on vaikea ennustaa. Sääntelykomplikaatiot tulevat hallitsemattomiksi varsinkin, jos geeniterapiaa ei voida paikantaa oikein kohdekudoksissa. Entsyymikorvaushoito voi myös olla ongelma, jos jotkin mutanttituotteet osoittautuvat dominoiviksi negatiivisiksi villin tyypin PLAG26: lle.
suurin osa INADIN mahdollisten hoitojen haasteista johtuu siitä, että kyseessä on erittäin harvinainen sairaus. Se, että INADILLA on tunnettu geneettinen etiologia, tarjoaa kuitenkin väyliä mahdollisille hoidoille. Lisäksi inadin onnistunut hoito saa harvinaislääkestatuksen ja kaiken sen saaman suojan, koska INAD on harvinainen sairaus. Näin ollen kaikista edellä mainituista haasteista huolimatta harvinaislääkeasema tarjoaa vahvan kannustimen harvinaisten sairauksien tutkijoille ja biotekniikkateollisuudelle, puhumattakaan siitä, että geneettinen syy on jo tiedossa.
geenin / emäksen muokkaus
vuodesta 2017 lähtien ihmisen PLA2G6-geenissä on havaittu ainakin 277 missense-mutaatiota (Lek et al., 2016). Vain pieni osa PLA2G6-mutaatioista sisältää kehyksensiirrot, indelit, nonsense-mutaatiot ja mutaatiot liitoskohdissa (Morgan et al., 2006). Näin kohdesolupopulaation geenin korjaaminen tai mutatoituneiden DNA-emästen korjaaminen on houkutteleva terapeuttinen keino. Vuosien varrella geenieditointiin on kehitetty useita työkaluja, ja CRISPR/Cas9-pohjaista genomieditointia pidetään uraauurtavana teknologiana, jonka kliininen tutkimus suunnitellaan vuonna 2018. Nämä teknologiat hyödyntävät DNA: ta sitova proteiini, joka voi myös cleave nauha tietyllä tavalla tehdä tilaa insertion uuden DNA-sekvenssin tai korjaus haitallisen DNA-emäksen (LaFountaine et al., 2015). CRISPR / Cas9-tekniikka on jo tuottanut hämmästyttäviä terapeuttisia tuloksia useissa prekliinisissä tautimalleissa (esim.Duchenen lihasdystrofia, maksan aineenvaihduntasairaudet jne.,) (Dai et al., 2016). Lisäksi, kasvava ymmärrys ihmisen vaihtelua aiheuttaa uudempia haasteita geeniterapian ja ajaa innovaatioita kohti todellista Personointi geenieditointi (Lessard et al., 2017; Scott ja Zhang, 2017). Uudet teknologiat, kuten vSLENDR, AAV-virus, ja CRISPR / Cas9-välitteinen tekniikka, jolla korvataan viallisia geenejä neuroneissa ja muissa hermoston soluissa, ajavat geenieditointiteknologioita uusille rajoille (Nishiyama et al., 2017).
johtopäätös
infantiili Neuroaksonaalinen dystrofia on vaikea neurodegeneratiivinen sairaus, johon liittyy tietty sairastuvuus ja kuolleisuus. Tämä harvinainen sairaus tarjoaa jännittävän mahdollisuuden uudistaa käytettävissä olevat seuraavan sukupolven hoidon ja tuottaa uudempia. Inad: n kliinisten diagnostisten kriteerien yhdenmukaisuuden odotetaan edistävän molekyylidiagnostiikan kehittämistä. Tämän edistyksen odotetaan johtavan siihen, että kehitämme kohtuuhintaisia hoitoja, jotka parantaisivat inad-potilaiden elämänlaatua määrällisesti ja hidastaisivat tai parantaisivat taudin etenemistä. Olemme korostaneet joitakin menestystarinoita Battenin taudista ja mukopolysakkaridooseista, jotka antavat meille inspiraation kysyä oikeita kysymyksiä INAD-hoidon toteuttamiseksi. Lisäksi kasvava virus-ja ei-virus lähestymistapoja CRISPR/Cas9 perustuva geenieditointi pitäisi myös avata uudempia terapeuttisia väyliä inad.
tekijöiden kannanotot
PB ja FA valmistelivat alustavan luonnoksen. SR ja AC muokkasivat ja lisäsivät käsikirjoitukseen lisää osia. DF, AP ja LP muokkasivat käsikirjoitusta.
Eturistiriitalausunto
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. A. (1995). Kalsiumista riippumattoman fosfolipaasi A2: n inhibitio estää arakidonihapon inkorporaation ja fosfolipidin remodelaation P388D1-makrofageissa. Proc. Natl. Acad. Sci. USA 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Tehokas keskushermoston kohdistaminen aikuisilla hiirillä intratekaalisella infuusiolla yksijuosteisella AAV9-GFP: llä neurologisten häiriöiden geeniterapiassa. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstract | CrossRef Full Text | Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 in vivo-geeniterapiaan: lupaus ja aitajuoksu. Mol. Ther. Nukleiinihapot 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstract | CrossRef Full Text | Google Scholar
Engel, L. A., Jing, Z., O ’ Brien, D. E., Sun, M., and Kotzbauer, P. T. (2010). PLA2G6: n katalyyttistä toimintaa heikentävät mutaatiot, jotka liittyvät infantiiliin neuroaksonaaliseen dystrofiaan, mutta eivät dystoniaan. PLoS yksi 5: e12897. doi: 10.1371 / lehti.pone.0012897
PubMed Abstrakti / poikkileikkaus koko teksti
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (1993). in PLA2G6-Associated Neurodegeneration, eds M. P. Adam, H. H. Ardinger, and R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). KESKUSHERMOSTOVAIKUTUS Fabryn tautiin: kliiniset ja kuvantamistutkimukset ennen 12 kuukauden entsyymikorvaushoitoa ja sen jälkeen. J. Perii. Metab. Tämä. 27, 229–240. doi: 10.1023 / B:BOLI.0000028794.04349.91
PubMed Abstract | CrossRef Full Text | Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Rather, M. I., Bhat, V., Gopinath, S., et al. (2016). Pla2g6: n geneettinen analyysi 22 intialaisessa perheessä, joissa on infantiili neuroaksonaalinen dystrofia, atyyppinen myöhäisen neuroaksonaalinen dystrofia ja dystonia parkinsonism complex. PLoS One 11: e0155605. doi: 10.1371 / lehti.pone.0155605
PubMed Abstract | CrossRef Full Text | Google Scholar
LaFountaine, J. S., Fathe, K., and Smyth, H. D. (2015). Toimitus ja terapeuttisia sovelluksia geenieditointiteknologioiden ZFNs. TALENs ja CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / J.ijpharm.2015.08.029
PubMed Abstract | CrossRef Full Text | Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291. doi: 10.1038 / nature19057
PubMed Abstract | CrossRef Full Text | Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Ihmisen geneettinen vaihtelu muuttaa CRISPR-Cas9 – spesifisyyttä sekä kohdentamisen että kohdentamisen suhteen terapeuttisesti kytketyssä lokuksessa. Proc. Natl. Acad. Sci. USA 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
PubMed Abstract | CrossRef Full Text | Google Scholar
Levi, S., and Finazzi, D. (2014). Neurodegeneraatio aivojen raudan kertymistä: patogeenisten mekanismien päivitys. Edessä. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Full Text / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Syklo-oksygenaasi-2: n ja kalsiumista riippumattoman fosfolipaasi A2: n mitokondriaalinen lokalisointi ihmisen syöpäsoluissa: implisiittinen vaikutus apoptoosin resistenssiin. Käyt.viim. Cell Res. 306, 75-84. doi: 10.1016 / j.yexcr.2005.01.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Macauley, S. L., and Sands, M. S. (2009). Lupaavaa keskushermostoon suunnattua entsyymikorvaushoitoa lysosomaalisten varastoitumissairauksien hoitoon. Käyt.viim. Neurol. 218, 5–8. doi: 10.1016 / J. expneurol.2009.03.040
PubMed Abstract | CrossRef Full Text | Google Scholar
Mehnaaz, L. (2016). Neurodegeneraatio aivojen raudan kertymistä (nbia) aiemmin Hallervorden – Spatz tauti. J. Assoc. Liikuntaa. Intia 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K., Morton, J. E. V., Gregory, A., Gissen, P., Sonek, S., et al. (2006). Fosfolipaasi A2: ta koodaava PLA2G6 mutatoituu neurodegeneratiivisissa häiriöissä, joissa on korkea aivorauta. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed Abstract | CrossRef Full Text | Google Scholar
Murphy, M. P. (2009). Miten mitokondriot tuottavat reaktiivisia happilajeja. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstract / CrossRef Full Text | Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C., and Faria, A. (2016). Neurodegeneration with brain iron accumulation: A case report. Dement. Neuropsykolia. 10, 160–164. doi: 10.1590 | S1980-5764-2016dn1002014
PubMed Abstract | CrossRef Full Text / Google Scholar
Nishiyama, J., Mikuni, T., and Yasuda, R. (2017). Virus-välitteinen genomin muokkaus kautta homology-suunnattu korjaus mitoottisten ja postmitoottisten solujen nisäkkäiden aivoissa. Neuroni 96, 755.e5-768.e5. doi: 10.1016 / J.neuron.2017.10.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., and Farooqui, A. A. (2005). Kalsiumista riippumattoman fosfolipaasi A2: n (iPLA2) jakautuminen apinan aivoissa. J. Neurosytoli. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
Pardridge, W. M. (2002). Lääke ja geeni kohdistetaan aivoihin molekylaarisilla troijalaisilla. Nat. Rev. Drug Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed Abstract / CrossRef Full Text | Google Scholar
Pardridge, W. M. (2005a). Veri-aivoesteen molekyylibiologia. Mol. Bioteknologiaa. 30, 57–70. doi: 10.1385 / MB:30: 1:057
CrossRef Full Text | Google Scholar
Pardridge, W. M. (2005b). Veri-aivoeste: pullonkaula aivojen lääkekehityksessä. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Kalsiumista riippumattomat fosfolipaasit A2 ja niiden rooli biologisissa prosesseissa ja sairauksissa. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstract | CrossRef Full Text | Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Taudin korjaus AAV-välitteisellä geeniterapialla mukopolysakkaridoosin tyypin IIID uudessa hiirimallissa. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg/ddx058
PubMed Abstract / CrossRef Full Text | Google Scholar
Scott, D. A., and Zhang, F. (2017). Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med. 23:1095. doi: 10,1038/nm.4377
PubMed Abstract | CrossRef Full Text | Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K., and Ma, Z. A. (2006). Kalsiumista riippumaton fosfolipaasi A2 paikallistaa ja suojaa mitokondrioita staurosporiinin apoptoottisen induktion aikana. J. Biol. Kemiaa. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstract | CrossRef Full Text | Google Scholar
Smith, A. J., Bainbridge, J. W., and Ali, R. R. (2012). Geenilisähoito periytyvien verkkokalvon dystrofioiden resessiivisiin muotoihin. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorrentino, N. C., D ’ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Erittäin erittyvä sulfamidaasi, joka on suunniteltu läpäisemään veri-aivoeste, korjaa tyypin IIIA mukopolysakkaridoosien hiirten aivovaurioita. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorrentino, N. C., and Fraldi, A. (2016). Aivojen kohdistaminen MPS-IIIA: ssa. Pediatr. Endokrinolia. Ilm. 13(Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., and Jones, S. S. (1997). Uusi sytosolikalsiumista riippumaton fosfolipaasi A2 sisältää kahdeksan ankyriiniaihetta. J. Biol. Kemiaa. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstract | CrossRef Full Text | Google Scholar
US Food and Drug Administration (2017). FDA hyväksyy Battenin taudin ensimmäisen hoidon. Saatavilla osoitteessa: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., and Gottlieb, R. A. (2002). Mitokondrion kalsiumista riippumattoman fosfolipaasi A2: n (iPLA2) esto vaimentaa mitokondrioiden fosfolipidihäviötä ja on kardioprotektiivinen. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstract / CrossRef Full Text | Google Scholar
Yu-Wai-Man, P. (2016). Geneettinen manipulointi perittyihin neurodegeneratiivisiin sairauksiin: myytti vai todellisuus? Br. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmoli-2015-308329
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et al. (2010). Haiman beetasolujen suojaaminen ryhmittäin fosfolipaasi A(2)-välitteisen mitokondrioiden kalvoperoksidaation korjauksen kautta. Endokrinologia 151, 3038-3048. doi: 10.1210 / Fi.2010-0016
PubMed Abstract | CrossRef Full Text / Google Scholar