Bevezetés
az infantilis neuroaxonális disztrófia (Inad) ismeretlen gyakoriságú autoszomális recesszív ritka neurodegeneratív betegség. A tünetek általában 6 hónapos és 3 éves kor között jelentkeznek. Ezt megelőzően a csecsemők normálisan fejlődnek. Az INAD első tünete lehet a normális fejlődési mérföldkövek elérésének lassulása vagy a fejlődési mérföldkövek regressziója (Ramanadham et al., 2015). A Trunk hypotonia, a strabismus és a nystagmus a betegség korai tünetei (Gregory et al., 2017). A betegség előrehaladása gyors, és előrehaladtával több megszerzett készség elveszik. Az izmok hamarosan hipotóniássá, később görcsössé válnak (Levi and Finazzi, 2014). Végül minden önkéntes izomkontroll elvész. Az izomgyengeség az etetés és a légzés nehézségeihez is vezethet. A nystagmus mellett néhány gyermek látásvesztést tapasztal. A kognitív funkciók fokozatosan elvesznek, és demencia alakul ki. Az élettartam általában 5-10 év (Gregory et al., 1993; Jardim et al., 2004; Macauley And Sands, 2009).
molekuláris patológia
az infantilis Neuroaxonális disztrófia a neurodegeneratív rendellenességek családjába tartozik, amely magában foglalja az atipikus késői neuroaxonális disztrófiát (ANAD) és a dystonia parkinsonizmus komplexet (DPC). Az INAD legtöbb esete homozigóta vagy összetett heterozigóta mutációkkal társul a PLA2G6 génben, amelyek befolyásolják fehérjetermékének katalitikus aktivitását (Engel et al., 2010). A PLA2G6 gén kalciumfüggetlen foszfolipáz A2 fehérjén (PLA2G6 vagy ipla2 65/88 kDa) keresztül kódol egy csoportot egy lipázzal és hét ankyrin ismétlődéstartalmú doménnel (Tang et al., 1997). A PLA2G6 hidrolizálja a foszfolipidek sn-2 Acil láncát, szabad zsírsavakat és lizofoszfolipideket hozva létre.
a mitokondriumok belső membránjában lévő foszfolipidek telítetlen zsírsavakban gazdagok az sn – 2 helyzetben, különösen a kardiolipinben (Seleznev et al., 2006). Ezek a telítetlen zsírsavak különösen érzékenyek a mitokondriumok által termelt bőséges reaktív oxigénfajokra (Murphy, 2009), ami peroxidált foszfolipideket eredményez a mitokondriumok belső membránjában. A PLA2G6 lokalizálódik a mitokondriumokban (Williams and Gottlieb, 2002; Liou et al., 2005) összhangban van a peroxidált zsírsavak hidrolízisének megnövekedett igényével a foszfolipidek SN-2 helyzetében, ami átalakult foszfolipidekhez vezet (Balsinde et al., 1995; Zhao et al., 2010). Ha a PLA2G6 hibás, a mitokondriumok belső membrán integritása megsérül. A PLA2G6 az axonra is lokalizálódik (Ong et al., 2005; Seleznev et al., 2006), ami a foszfolipid átalakítás iránti megnövekedett helyi igényt jelzi ott is. Az ilyen felhalmozódás megnyilvánulása az agyban egyedülálló a kulcsfontosságú agyterületeken, például a bazális ganglionokon, amelyek ugyanazon mögöttes molekuláris patogenezis különböző neveit eredményezték PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
a pla2g6 kiütéses egerek neuronjainak ultrastrukturális elemzése összhangban áll ezzel a molekuláris patológiával. Mitokondriumokat elágazó és tubuláris cristae-val, mitokondriumokat degenerált cristae-val, axonokat citoszkeleton összeomlással és részleges membránvesztést figyeltek meg az axon terminálokon (Beck et al., 2011). Mikroszkopikus szinten ezek a jellemzők axonális duzzanatokként és szferoid testekként jelennek meg a központi vagy perifériás idegrendszer pre-szinaptikus terminálisaiban (1.ábra).
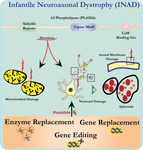
1. ábra. Az infantilis neuroaxonális disztrófia (INAD) egy neurodegeneratív rendellenesség, amely a PLA2G6 gén mutációival kapcsolatos. A PLA2G6 különböző mutációi diszfunkcionális A2 foszfolipázhoz vezetnek, ami mitokondriális és axonális membránhibákhoz vezet. Ezek a hibák neuronális károsodást okoznak axonális duzzanatokként és a pre-szinaptikus szferoidok felhalmozódásaként. Enzimpótlás a funkciók helyreállításához, génpótlás vagy szerkesztés a hibás PLA2G6 kijavításához javasolt terápiás stratégiák.
molekuláris diagnosztika és ritka betegségben szenvedő betegek felhatalmazása
a specifikus klinikai, elektrofiziológiai és képalkotó funkcióktól eltekintve, a következő generációs szekvenálás elérhetősége előtt az inad megerősítésének diagnosztikai kritériumai voltak a központi vagy perifériás idegrendszer pre-szinaptikus terminálisaiban axonális duzzanatokat és szferoid testeket mutató bőrbiopsziák (Gregory et al., 2017; Iodice et al., 2017). Gyakran több biopsziára volt szükség a diagnózis megerősítéséhez. A családok általában sok évet vártak a diagnózisra. A gén-és genomszekvenálás költségeinek csökkenésével, a diagnosztizálatlan neurológiai betegségek diagnosztikai laboratóriumokkal történő célzott génpanel-tesztelésének elérhetőségével és az orvosok növekvő tudatosságával a genetikai diagnosztika elérhetőségével a családok gyorsabban kapják meg a diagnózist; néha az első tünet megjelenésétől számított egy éven belül. Ezekben a családokban a gyerekek még fiatalok, és a családok motiváltak arra, hogy a tudósokkal együttműködve megtalálják gyermekeik betegségének kezelését. A kutatási folyamat finanszírozása érdekében az INAD betegek szüleinek egy csoportja megalapította az INADcure Alapítványt. Az alapítvány jelentős forrásokat gyűjtött a kutatáshoz, és együttműködik a ritka genomikai Intézettel, hogy irányítsa őket a kutatási támogatások odaítélésében. Érdekes módon egy 2016-os genetikai elemzés 22 indiai családok INAD, ANAD, és DPC megállapította, hogy 10/22 család (45,45%) hiányzott mutációk a PLA2G6 gén kódoló régióban (Kapoor et al., 2016). A pla2g6 kódoló régiójában a káros mutációk azonosításának elmulasztása rávilágít arra, hogy a jövőbeni molekuláris diagnosztikai erőfeszítésekhez teljes génszekvenálásra lenne szükség a PLA2G6 gén intronikus és szabályozó régióinak mutációinak azonosításához INAD érintett betegeknél. Továbbá az olyan esetek, amikor a PLA2G6-hoz kapcsolódó mutációkat nem figyelték meg, arra utalnak, hogy a betegség oka a PLA2G-től eltérő gének mutációja lehet, amelyet meg kell vizsgálni.
lehetséges terápiák
egy olyan ritka betegség esetében, mint az inad, amely hibás enzimet tartalmaz, a legtöbb terápia enzimpótlás, génpótlás vagy génkorrekció. Ha az enzimhiányt recesszív genetikai hiba okozza, feltételezzük, hogy az enzimpótlás vagy kiegészítés kijavíthatja a problémát (Smith et al., 2012; Yu-Wai-Man, 2016). Azonban a kísérletek annak bizonyítására, hogy a megfelelő gént vagy enzimet biztosító terápiák megmentik az INAD fenotípust, még nem végeztek és teszteltek.
enzimpótló terápia (ERT)
mivel az inad-ban az agy az elsődleges szerv, az inad enzimpótló terápiájához nagy valószínűséggel az enzim agyba történő infúziója szükséges. 2017-ben a Biomarin megkapta az FDA jóváhagyását a tripeptidil-peptidáz 1 (cerliponáz alfa) kezelésére a Batten-betegség, a tripeptidil-peptidáz (TPP1) hiánya lizoszomális enzim (USA Food Drug Administration, 2017). Tripeptidil-peptidáz 1 (cerliponáz alfa; 59 kDa) az első ERT, amelyet közvetlenül az agy cerebrospinális folyadékába (CSF) adnak be. A CSF-be beadható egyéb ERT-gyógyszerek klinikai vizsgálatokban vannak (Jardim et al., 2004; Macauley And Sands, 2009).
a tripeptidil-peptidáz 1 (cerliponáz alfa) hatékonysága a Batten-betegségben szenvedő betegek járóképességén azt mutatja, hogy az agyat érintő betegségek enzimpótló terápiája elméletileg lehetséges. A helyettesítő enzim célzása az INAD mitokondriumaiba nagyobb kihívást jelent, mint a lizoszomális célzás, amely már hosszú ideje sikeres volt az IV ERT által, mint a Gaucher-kór esetében. Ezért még mindig sok, az INAD-ra jellemző biológiai kérdés van, amelyeket meg kell oldani:
• szükség van-e az INAD terápiájára olyan mechanizmusokra, amelyek esetleg pla2g6-ot szállíthatnak a sejtbe?
további probléma, hogy az enzimpótló kezelés megköveteli az intracerebroventricularis katéter kezdeti elhelyezését és gyakori infúziókat. A katéter elhelyezése érzéstelenítést igényel, az infúziók pedig érzéstelenítést igényelhetnek a betegek együttműködésétől függően. Az inad-ben szenvedő gyermekek gyermekneurológusainak és szüleinek aggodalmainak kezelése az anesztézia alkalmazásával kapcsolatban inad-betegeknél megfelelő információkkal további erőfeszítéseket igényel.
génterápia/Génpótlás
az emberi PLA2G gén körülbelül 70 kb, 17 exonnal és 2 Alternatív exonnal. A leghosszabb fehérje-kódoló transzkriptum azonban csak 3,3 kb, a fehérje kódoló szekvencia pedig alig több mint 2,4 kb, amelyet könnyen be lehet csomagolni egy vírusvektor rakományba. Először egy másik lizoszomális tárolási rendellenességet fedezünk fel a központi idegrendszerben, amelyek preklinikai adatokkal rendelkeznek, majd összehasonlíthatók az INAD-val. Az ERT preklinikai adatainak hiánya az INAD-ban jelentős ismerethiány a területen, ezért elengedhetetlen, hogy megfigyeljük a központi idegrendszeri rendellenességek egyéb preklinikai modelljeit, amelyek génterápiás vizsgálatokon mennek keresztül. Korábban egereken végzett preklinikai vizsgálat foglalkozott a lizoszomális tárolási rendellenesség GÉNTERÁPIÁJÁVAL IIIa típusú mukopoliszacharidózisok (MPS-IIIA) a szulfamidáz gén helyes változatának vírusvektorba csomagolásával (Sorrentino et al., 2013). A tanulmány kihasználta a vér-agy gát (BBB) növekvő megértését, amely aktívan szabályozza a nagy molekulák szállítását a vérből a központi idegrendszerbe egy transzcitózisnak nevezett folyamat révén (Pardridge, 2005b; Sorrentino and Fraldi, 2016).
a Transzcitózis magában foglalja a ligandumok endocitózisát a luminalis oldalon, ligandum-specifikus receptorok (pl. inzulinreceptor, transzferrin receptorés alacsony sűrűségű lipoprotein receptor stb.,) a kapilláris endotheliumon dúsítva (Pardridge, 2005a). Ezt követi az endocytosed rakomány mozgása az endothelium citoplazmán keresztül, végül exocitózis az abluminalis (agy) oldalon, így hatékonyan szállítja a rakományt a BBB-n keresztül (Pardridge, 2002). Az MPS-IIIA preklinikai vizsgálat egy kiméra szulfamidáz fehérjét gyártott, amely BBB-kötő domént tartalmazott az apolipoprotein B-ből, hogy megkönnyítse az endothelium felvételét, valamint az iduronát-2-szulfatáz szignálpeptidjét, hogy elősegítse a hatékony exocitózist a BBB abluminális oldala felé (Sorrentino et al., 2013). Egy kiméra szulfamidázt kódoló vírusvektor rakományt ezután a májat célzó 2/8-as szerotípusú adeno-asszociált vírusra (AAV) töltöttünk (Sorrentino and Fraldi, 2016). Így a máj belső gyárként szolgált, amely állandó kiméra-szulfamidáz-ellátást biztosított, ami az agy szulfamidáz aktivitásának 10-15% – os növekedését eredményezte még 7 hónappal a máj génterápiája után is. Az agyi szulfamidáz aktivitás szintjének növekedése az agy patológiájának és viselkedési eredményeinek számszerűsíthető javulásához vezetett az MPS-IIIA egérmodelljében (Sorrentino et al., 2013).
ha hasonló vizsgálatot végeznek az INAD-val kapcsolatban, az számos kritikus kérdésre válaszolna az inad terápia enzimpótlásával kapcsolatban. Alternatív megoldásként az aav9 alapú génterápiás termékek vaszkuláris vagy CSF-en belüli beadása közvetlenül megcélozhatja a központi idegrendszert (Bey et al., 2017; Roca et al., 2017). Azonban a többszörös mutációk, amelyek az INAD-betegeknél a PLAG26 génjükön vannak, egyedülálló kihívásokat jelentenek a génterápia számára. Bár a PLA2G6 gén mérete 2-3 kb nem jelenthet problémát a vírusvektorba történő beillesztésében, a pla2g6 korrekciójának szabályozási szövődményeit azonban nehéz megjósolni. A szabályozási szövődmények ellenőrizhetetlenné válnak, különösen akkor, ha a génterápia nem lokalizálható megfelelően a célszövetekben. Az enzimpótló terápia akkor is problémát jelenthet, ha egyes mutáns termékek domináns negatívnak bizonyulnak a vad típusú PLAG26-ra.
az INAD lehetséges terápiáinak legtöbb kihívása abból adódik, hogy rendkívül ritka betegségről van szó. Az a tény azonban, hogy az INAD ismert genetikai etiológiával rendelkezik, lehetőséget nyújt a lehetséges terápiákra. Ezenkívül az INAD sikeres terápiája Ritka Betegségek Gyógyszer státusát és minden védelmet kap, mivel az INAD ritka betegség. Így a fent említett kihívások ellenére a ritka betegségek gyógyszereinek státusza erős ösztönzést nyújt a ritka betegségek kutatóinak és a biotechnológiai iparnak, nem is beszélve arról, hogy a genetikai ok már ismert.
gén / bázis Szerkesztés
2017-től legalább 277 missense mutációt figyeltek meg az emberi PLA2G6 génben (Lek et al., 2016). A PLA2G6 mutációknak csak egy kis hányada tartalmazza a kereteltolódásokat, az indeleket, a nonszensz mutációkat és a splice helyek mutációit (Morgan et al., 2006). Így a gén korrekciója a célsejtpopulációban vagy a mutált DNS-bázisok korrekciója vonzó terápiás út. Az évek során számos eszközt fejlesztettek ki a génszerkesztéshez, és a CRISPR/Cas9-alapú genomszerkesztést úttörő technológiának tekintik egy 2018-ban tervezett klinikai vizsgálattal. Ezek a technológiák olyan DNS-kötő fehérjét használnak, amely a szálat is meghatározott módon hasíthatja, hogy helyet biztosítson egy új DNS-szekvencia beillesztéséhez vagy a káros DNS-bázis korrekciójához (LaFountaine et al., 2015). A CRISPR / Cas9 technológia már meghökkentő terápiás eredményeket hozott számos preklinikai betegségmodellben(pl.,) (Dai et al., 2016). Ezenkívül az emberi variáció növekvő megértése újabb kihívásokat jelent a génterápia számára, és az innovációt a génszerkesztés valódi személyre szabása felé irányítja (Lessard et al., 2017; Scott és Zhang, 2017). Az olyan új technológiák, mint a vSLENDR, az AAV vírus és a CRISPR/Cas9 által közvetített technológia a neuronok és más idegrendszeri sejtek hibás génjeinek helyettesítésére, a génszerkesztő technológiákat új határok felé tolják (Nishiyama et al., 2017).
következtetés
az infantilis Neuroaxonális dystrophia súlyos neurodegeneratív betegség, bizonyos morbiditással és mortalitással. Ez a ritka betegség izgalmas lehetőséget kínál a következő generációs terápia elérhető módjainak újraértékelésére és újabbak létrehozására. Az INAD klinikai diagnosztikai kritériumainak kialakuló kongruenciája várhatóan lendületet ad a fokozott molekuláris diagnosztika felé. Ez az előrelépés várhatóan megfizethető terápiák kifejlesztéséhez vezet, amelyek számszerűsíthető javulást biztosítanának az INAD-betegek életminőségében, késleltetve vagy enyhítve a betegség progresszióját. Kiemeltünk néhány sikertörténetet a Batten-kórban és a mukopoliszacharidózisokban, amelyek inspirációt nyújtanak számunkra, hogy feltegyük a megfelelő kérdéseket, hogy az INAD terápia valósággá váljon. Ezenkívül a CRISPR/Cas9 alapú génszerkesztés növekvő vírusos és nem vírusos megközelítéseinek újabb terápiás utakat kell nyitniuk az INAD számára.
szerzői hozzájárulások
a PB és az FA előzetes tervezetet készített. Az SR és az AC szerkesztette és bővítette a kéziratot. DF, AP és LP szerkesztette a kéziratot.
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K. és Dennis, E. A. (1995). A kalciumfüggetlen foszfolipáz A2 gátlása megakadályozza az arachidonsav beépülését és a foszfolipid átalakulását a p388d1 makrofágokban. Proc. NAT. Acad. Sci. U. S. A. 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Hatékony központi idegrendszeri célzás felnőtt egerekben egyszálú aav9-GFP intrathecalis infúzióval neurológiai rendellenességek génterápiájához. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 in vivo génterápia: ígéret és akadályok. Mol. Ott. Nukleinsavak 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
Engel, L. A., Jing, Z., O ‘ Brien, D. E., Sun, M. és Kotzbauer, P. T. (2010). A PLA2G6 katalitikus funkcióját az infantilis neuroaxonális disztrófiával kapcsolatos mutációk károsítják, de nem a dystonia-parkinsonizmus. PLoS egy 5: e12897. doi: 10.1371 / folyóirat.pone.0012897
PubMed absztrakt / CrossRef teljes szöveg
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P. és Hayflick, S. J. (1993). a PLA2G6-hoz kapcsolódó Neurodegenerációban eds M. P. Adam, H. H. Ardinger és R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Központi idegrendszeri érintettség Fabry-kórban: klinikai és képalkotó vizsgálatok 12 hónapos enzimpótló kezelés előtt és után. J. Örökölni. Metabolit. Dis. 27, 229–240. doi: 10.1023 / B:BOLI.0000028794.04349.91
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Kapoor, S., Shah, M. H., Singh, N., inkább M. I., Bhat, V., Gopinath, S., et al. (2016). Pla2g6 genetikai analízise 22 indiai családban infantilis neuroaxonális disztrófiában, atipikus késői neuroaxonális disztrófiában és dystonia parkinsonism komplexben. PLoS egy 11: e0155605. doi: 10.1371 / folyóirat.pone.0155605
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
LaFountaine, J. S., Fathe, K. és Smyth, H. D. (2015). A zfns génszerkesztő technológiák szállítása és terápiás alkalmazása. TALENs és CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). A fehérjét kódoló genetikai variáció elemzése 60 706 emberben. Természet 536, 285-291. doi: 10.1038 / nature19057
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Az emberi genetikai variáció megváltoztatja a CRISPR – Cas9 be-és off-célzási specifitását a terápiásán érintett lokuszokban. Proc. NAT. Acad. Sci. U. S. A. 14, E11257–E11266. doi: 10.1073 / pnas.1714640114
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Levi, S. és Finazzi, D. (2014). Neurodegeneráció agyi vas felhalmozódásával: a patogén mechanizmusok frissítése. Elöl. Pharmacol. 5:99. doi: 10.3389 / ffar.2014.00099
CrossRef teljes szöveg / Google Scholar
J. J., J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). A ciklooxigenáz-2 és a kalciumfüggetlen foszfolipáz A2 mitokondriális lokalizációja humán rákos sejtekben: az apoptózis-rezisztencia következménye. Felh. 306-Os, 75-84-Es Cella. doi: 10.1016 / j. yexcr.2005.01.011
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Macauley, S. L. és Sands, M. S. (2009). Ígéretes központi idegrendszeri enzimpótló terápia lizoszomális tárolási betegségek esetén. Felh. Neurol. 218, 5–8. doi: 10.1016 / j. expneurol.2009.03.040
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
Mehnaaz, L. (2016). Neurodegeneráció agyi vas felhalmozódásával (NBIA) korábban Hallervorden – Spatz betegség. J. Assoc. Phys. India 64:132.
Google Tudós
Morgan, N. V., Westaway, S. K., Morton, J. E. V., Gergely, A., Gissen, P., Sonek, S. et al. (2006). PLA2G6, kódoló foszfolipáz A2, mutálódik neurodegeneratív rendellenességek magas agyi vas. Nat. Genet. 38, 752–754. doi: 10.1038 | ng1826
PubMed absztrakt | CrossRef teljes szöveg / Google Tudós
Murphy, M. P. (2009). Hogyan termelnek a mitokondriumok reaktív oxigénfajokat. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Nassif, D., Pereira, J. S., Spitz, M., capit 6.és Faria, A. (2016). Neurodegeneráció agyi vas felhalmozódásával: esettanulmány. Dement. Neuropszichol. 10, 160–164. doi: 10.1590 / S1980-5764-2016dn1002014
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Nishiyama, J., Mikuni, T. és Yasuda, R. (2017). Vírus által közvetített genomszerkesztés homológia által irányított javítással az emlősök agyának mitotikus és posztmitotikus sejtjeiben. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j. neuron.2017.10.004
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F. és Farooqui, A. A. (2005). A kalciumfüggetlen foszfolipáz A2 (iPLA2) eloszlása a majom agyában. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Pardridge, W. M. (2002). Drog és gén célzás az agy molekuláris trójai falovakkal. Nat. Drug Discov Tiszteletes. 1, 131–139. doi: 10.1038 / nrd725
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Pardridge, W. M. (2005a). A vér-agy gát molekuláris biológiája. Mol. Biotechnol. 30, 57–70. doi: 10.1385 / MB:30: 1:057
CrossRef teljes szöveg / Google Scholar
Pardridge, W. M. (2005b). A vér-agy gát: szűk keresztmetszet az agyi gyógyszerfejlesztésben. NeuroRx 2, 3-14.
Google Scholar
R. N., R. N., Hancock, W. D., Lei, X., et al. (2015). Kalciumfüggetlen A2 foszfolipázok és szerepük a biológiai folyamatokban és betegségekben. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Roca, C., Motas, S., Márc., Ribera, A., S., S., V., S., X., et al. (2017). A betegség korrekciója AAV által közvetített génterápiával a IIID típusú mukopoliszacharidózis új egérmodelljében. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg / ddx058
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Scott, D. A. és Zhang, F. (2017). Az emberi genetikai variáció következményei a CRISPR-alapú terápiás genomszerkesztésben. Nat. Med. 23:1095. doi: 10,1038 / nm.4377
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Szeleznyev, K., Zhao, C., Zhang, X. H., Song, K. és Ma, Z. A. (2006). A kalciumfüggetlen foszfolipáz A2 lokalizálja és védi a mitokondriumokat a sztauroszporin apoptotikus indukciója során. J. Biol. Kémia. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Smith, A. J., Bainbridge, J. W. és Ali, R. R. (2012). Génpótló terápia az öröklött retinális dystrophiák recesszív formáira. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
Sorrentino, N. C., D ‘ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Egy erősen szekretált szulfamidáz, amelyet a vér-agy gáton való átjutásra terveztek, korrigálja az egerek agyi elváltozásait a IIIA típusú mukopoliszacharidózisokkal. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Sorrentino, N. C. és Fraldi, A. (2016). Agyi célzás az MPS-IIIA – ban. Pediatr. Endokrinol. Jel. 13 (Kiegészítés. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J. és Jones, S. S. (1997). Egy új citoszolos kalciumfüggetlen foszfolipáz A2 nyolc ankyrin motívumot tartalmaz. J. Biol. Kémia. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Amerikai Élelmiszer-és Gyógyszerügyi Hivatal (2017). Az FDA jóváhagyja a Batten-betegség egyik formájának első kezelését. Elérhető itt:: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D. és Gottlieb, R. A. (2002). A mitokondriális kalciumfüggetlen foszfolipáz A2 (iPLA2) gátlása csökkenti a mitokondriális foszfolipid veszteséget és kardioprotektív. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Yu-Wai-Man, P. (2016). Genetikai manipuláció öröklött neurodegeneratív betegségek esetén: mítosz vagy valóság? Br. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et al. (2010). A hasnyálmirigy béta-sejtjeinek védelme csoportonként a mitokondriális membrán peroxidációjának foszfolipáz A(2) által közvetített javításán keresztül. Endokrinológia 151, 3038-3048. doi: 10.1210 / hu.2010-0016
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós