Innledning
Infantile neuroaxonal dystrophy (INAD) er en autosomal recessiv sjelden nevrodegenerativ sykdom med ukjent frekvens. Utbruddet av symptomer oppstår vanligvis mellom 6 måneder og 3 år. Før den tiden utvikler spedbarn normalt. DET første symptomet PÅ INAD kan være bremsing av oppnåelsen av normale utviklingsmilepæler eller regresjon i utviklingsmilepæler (Ramanadham et al., 2015). Trunk hypotoni, strabismus og nystagmus er tidlige symptomer på sykdommen (Gregory et al., 2017). Progresjonen av sykdommen er rask, og når den utvikler seg, går mer oppnådde ferdigheter tapt. Muskler blir snart hypotoniske og blir senere spastiske (Levi Og Finazzi, 2014). Til slutt går all frivillig muskelkontroll tapt. Muskel svakhet kan også føre til vanskeligheter med å mate og puste. I tillegg til nystagmus opplever noen barn synstap. Kognitive funksjoner går gradvis tapt og demens utvikler seg. Levetiden er vanligvis 5 til 10 år (Gregory et al., 1993; Jardim et al.(2004; Macauley Og Sands, 2009).
Molekylær Patologi
Infantil Neuroaksonal Dystrofi tilhører en familie av nevrodegenerative lidelser som inkluderer atypisk sen debut neuroaksonal dystrofi (ANAD) og dystoni Parkinsonisme kompleks (DPC). De fleste tilfeller AV INAD er forbundet med homozygote eller sammensatte heterozygote mutasjoner I PLA2G6-genet som påvirker den katalytiske aktiviteten til proteinproduktet (Engel et al., 2010). PLA2G6-genet koder for en gruppe via kalsiumuavhengig fosfolipase A2-protein (PLA2G6 eller IPLA2ß, ∼85/88 kDa) med en lipase og syv ankyrin-repeterende domener (Tang et al., 1997). PLA2G6 hydrolyserer sn-2 acyl-kjeden av fosfolipider, genererer frie fettsyrer og lysofosfolipider.
Fosfolipider i mitokondriens indre membran er rike på umettede fettsyrer i sn-2-posisjonen, spesielt kardiolipin (Seleznev et al., 2006). Disse umettede fettsyrene er spesielt sårbare for de rike reaktive oksygenartene produsert av mitokondriene (Murphy, 2009) som resulterer i peroksiderte fosfolipider i mitokondriens indre membran. PLA2G6 lokaliserer til mitokondriene (Williams og Gottlieb, 2002; Liou et al., 2005) i samsvar med økt etterspørsel etter hydrolyse av de peroxidiserte fettsyrene i sn-2-posisjonen til fosfolipider som fører til ombygde fosfolipider (Balsinde et al., 1995; Zhao et al., 2010). NÅR PLA2G6 er defekt, er mitokondriens indre membranintegritet skadet. PLA2G6 lokaliserer også til axon (Ong et al., 2005; Seleznev et al., 2006) indikerer en økt lokalisert etterspørsel etter fosfolipid remodeling der også. Manifestasjonen av slik akkumulering i hjernen er unik for viktige hjerneområder, som basalganglia som resulterte i forskjellige navn for den samme underliggende molekylære patogenesen som involverer PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
Ultrastrukturanalyse av nevroner I PLA2G6 knockout-mus er i samsvar med denne molekylære patologien. Mitokondrier med forgrening og rørformet cristae, mitokondrier med degenerert cristae, axoner med cytoskelettkollaps og delvis membrantap ved axonterminaler er observert (Beck et al., 2011). På mikroskopisk nivå opptrer disse trekkene som aksonale hevelser og sfæroide legemer i pre-synaptiske terminaler (Figur 1) i det sentrale eller perifere nervesystemet.
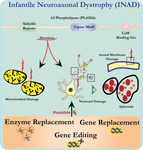
Figur 1. Infantil neuroaxonal dystrofi (INAD) er en nevrodegenerativ lidelse relatert til mutasjoner I PLA2G6 genet. ULIKE mutasjoner AV PLA2G6 fører til dysfunksjonell a2 fosfolipase som fører til mitokondrielle og aksonale membranfeil. Disse feilene forårsaker nevronskader visualisert som aksonale hevelser og akkumulering av pre-synaptiske sfæroider. Enzym erstatning for å gjenopprette funksjoner, gen erstatning eller redigering for å korrigere den defekte PLA2G6 er foreslått terapeutiske strategier.
Molekylær Diagnostikk Og Sjeldne Sykdom Pasient Empowerment
Bortsett fra spesifikke kliniske, elektrofysiologiske, og imaging funksjoner, før tilgjengeligheten av neste generasjons sekvensering, hud biopsier viser aksonal hevelser og sfæroide organer i pre-synaptiske terminaler i det sentrale eller perifere nervesystemet var de diagnostiske kriteriene for bekreftelse AV INAD (Gregory et al ., 2017; Iodice et al., 2017). Ofte var det nødvendig med flere biopsier for å bekrefte diagnosen. Familier ventet i mange år på en diagnose. Med den avtagende kostnaden for gen-og genomsekvensering, tilgjengeligheten av målrettet genpaneltesting med diagnostiske laboratorier for udiagnostiserte nevrologiske sykdommer, og den økende bevisstheten til leger om tilgjengeligheten av genetisk diagnostikk, mottar familier diagnosen raskere; noen ganger innen et år etter det første symptomet som vises. Barna i disse familiene er fortsatt unge og familiene er motivert til å samarbeide med forskere for å finne en behandling for sine barns sykdom. For å finansiere forskningsprosessen dannet En gruppe foreldre TIL inad-pasienter INADcure Foundation. Stiftelsen har reist betydelige midler til forskning og samarbeider Med Rare Genomics Institute for å veilede dem i tildeling av forskningsstipendier. Interessant, en 2016 genetisk analyse av 22 Indiske familier MED INAD, ANAD og DPC fant at 10 / 22 familier (45.45%) manglet mutasjoner I PLA2G6-genkodingsområdet (Kapoor et al., 2016). Unnlatelse av å identifisere skadelige mutasjoner I KODINGSOMRÅDET TIL PLA2G6 fremhever at fremtidig molekylær diagnostisk innsats vil kreve hel gensekvensering for å identifisere mutasjoner i introniske og regulatoriske regioner av PLA2G6-genet hos INAD-berørte pasienter. VIDERE ANTYDER INAD-tilfeller der PLA2G6-assosierte mutasjoner ikke observeres at årsaken til sykdommen kan skyldes mutasjon i andre gener enn PLA2G, som må utforskes.
Mulige Terapier
De fleste terapiene som vurderes for en sjelden sykdom som INAD involverer et defekt enzym, er enzymutskifting, genutskifting eller genkorreksjon. Når et enzym mangel er forårsaket av en recessiv genetisk defekt, antas det at enzym erstatning eller tilskudd kan løse problemet (Smith et al., 2012; Yu-Wai-Man, 2016). Imidlertid er eksperimenter for å bevise at terapier som gir riktig gen eller enzym, vil redde inad-fenotypen, ennå ikke utført og testet.
ENZYME Replacement Therapy (ERT)
siden hjernen er det primære organet som påvirkes i INAD, vil enzymbehandling for INAD mest sannsynlig kreve infusjon av enzymet i hjernen. I 2017 Mottok Biomarin FDA-godkjenning for tripeptidylpeptidase 1 (cerliponase alfa) som behandling for den underliggende årsaken Til Batten sykdom, mangelen på tripeptidylpeptidase (TPP1) et lysosomalt enzym (US Food Drug Administration, 2017). Tripeptidylpeptidase 1 (cerliponase alfa; ∼59 kDa) er det første ERT som administreres direkte i cerebrospinalvæsken (CSF) i hjernen. ANDRE ERT-legemidler som kan administreres i CSF er i kliniske studier (Jardim et al.(2004; Macauley Og Sands, 2009).
effekten av tripeptidylpeptidase 1 (cerliponase alfa) på Gangevnen til Batten sykdomspasienter viser at enzymutskiftningsterapi for sykdommer som påvirker hjernen er teoretisk mulig. Målretting av erstatningsenzym i mitokondriene for INAD vil være mer utfordrende enn lysosomal målretting som ALLEREDE hadde vært vellykket AV IV ERT i lang tid som I Tilfelle Av Gaucher sykdom. Derfor er det fortsatt mange biologiske problemer som er spesifikke FOR INAD som må løses:
* trenger terapi FOR INAD mekanismer som muligens kan transportere PLA2G6 inn i cellen?
et ekstra problem er at enzymutskiftningsterapi krever den første plasseringen av et intracerebroventrikulært kateter og hyppige infusjoner. Plasseringen av kateteret krever anestesi og infusjoner kan kreve anestesi avhengig av pasientens samarbeid. Adressering bekymringene til pediatriske nevrologer og foreldre til BARN MED INAD med hensyn til bruk av anestesi PÅ INAD pasienter med riktig informasjon vil kreve ytterligere innsats.
Genterapi/Genutskifting
DET humane PLA2G-genet er rundt 70 kb med 17 eksoner og 2 alternative eksoner. Den lengste proteinkodende transkripsjonen er imidlertid bare 3,3 kb, og proteinkodingssekvensen er litt over 2,4 kb, som lett kan pakkes inn i en viral vektorlast. Vi vil først undersøke en annen lysosomal lagringsforstyrrelse i CNS som har prekliniske data som skal sammenlignes med INAD. Mangelen på prekliniske data AV ERT i INAD er et betydelig kunnskapsgap på feltet, og derfor er det avgjørende at vi observerer andre prekliniske modeller AV CNS-lidelser som gjennomgår genterapistudier. Tidligere behandlet en preklinisk studie hos mus genterapi for lysosomal lagringsforstyrrelse mukopolysakkaridoser type iiia (MPS-IIIA) ved å pakke den riktige versjonen av sulfamidasegenet inne i en viral vektor (Sorrentino et al., 2013). Studien tok fordel av den økende forståelsen av blodhjernebarrieren (BBB) som aktivt regulerer transport av store molekyler fra blod til CNS ved en prosess som kalles transcytose (Pardridge, 2005b; Sorrentino og Fraldi, 2016).
Transcytose innebærer endocytose av ligander på luminalsiden, mediert av ligand-spesifikke reseptorer(f. eks. insulinreseptor, transferrinreseptor og lavdensitets-lipoproteinreseptor, etc.,) beriket på kapillærendotelet (Pardridge, 2005a). Dette etterfølges av bevegelse av endocytosed last gjennom endotelet cytoplasma og til slutt eksocytose på abluminal (hjerne) side, og dermed effektivt levere lasten OVER BBB (Pardridge, 2002). Mps-iiia-preklinisk studie produserte et kimært sulfamidaseprotein som inneholdt ET BBB-bindende domene fra apolipoprotein B for å lette opptaket av endotelet og også et signalpeptid fra iduronat-2-sulfatase for å hjelpe en effektiv eksocytose mot DEN abluminale siden AV BBB (Sorrentino et al., 2013). En viral vektorlast som koder for en kimær sulfamidase ble deretter lastet på et adeno-assosiert virus (aav) serotype 2/8 rettet mot leveren (Sorrentino Og Fraldi, 2016). Således fungerte leveren som en intern fabrikk som ga en konstant tilførsel av kimær-sulfamidase, noe som resulterte i en 10-15% økning i hjernesulfamidaseaktivitet selv 7 måneder etter levergenterapi. Denne økningen i hjernesulfamidaseaktivitetsnivåer førte til kvantifiserbar forbedring i hjernepatologi og atferdsutfall i musemodellen AV MPS-Iiia (Sorrentino et al., 2013).
hvis EN lignende studie utføres FOR INAD, vil DEN svare på flere kritiske spørsmål om enzymutskifting for INAD-terapi. Alternativt kan intra-vaskulær eller intra-CSF administrasjon av aav9-baserte genterapiprodukter direkte målrette CNS (Bey et al., 2017; Jørgen et al., 2017). Imidlertid gir de flere mutasjonene SOM INAD-pasienter har på DERES PLAG26-gen unike utfordringer for genterapi. Selv om STØRRELSEN PÅ PLA2G6-genet på 2-3 kb ikke bør utgjøre et problem for innføring i en viral vektor, er de regulatoriske komplikasjonene ved å korrigere PLA2G6 vanskelig å forutsi. Regulatoriske komplikasjoner blir ukontrollable, spesielt hvis genterapien ikke kan lokaliseres riktig i målvevet. Enzymerstatningsterapi kan også være et problem hvis noen mutantprodukter viser seg å være dominerende negativ TIL VILLTYPE PLAG26.
de fleste utfordringene med mulige terapier for INAD kommer fra det faktum at DET er en ultra sjelden sykdom. MEN DET FAKTUM AT INAD har en kjent genetisk etiologi gir veier for mulige terapier. Videre vil enhver vellykket behandling for INAD få foreldreløs narkotikastatus og all beskyttelse den får fordi INAD er en sjelden sykdom. Dermed til tross for alle de nevnte utfordringene, gir orphan drug status et sterkt incitament til sjeldne sykdomsforskere og bioteknologiindustrien, for ikke å nevne når den genetiske årsaken allerede er kjent.
Gen / Base Redigering
Fra og med 2017 er det observert minst 277 missense mutasjoner i DET humane PLA2G6-genet (Lek et al., 2016). BARE en liten andel AV PLA2G6-mutasjoner inkluderer rammeskift, indels, nonsensmutasjoner og mutasjoner i spleisestedene (Morgan et al., 2006). Således korrigere genet i målcellepopulasjonen eller korrigere de muterte DNA-basene er en attraktiv terapeutisk avenue. GJENNOM årene har flere verktøy blitt utviklet for genredigering, OG CRISPR / Cas9-basert genomredigering blir hyllet som en banebrytende teknologi med en klinisk studie planlagt i 2018. Disse teknologiene bruker ET DNA-bindende protein som også kan spalte strengen på en bestemt måte for å gi plass til innføring av en ny DNA-sekvens eller korreksjon av den skadelige DNA-basen (LaFountaine et al., 2015). CRISPR / Cas9-teknologien har allerede gitt forbløffende terapeutiske resultater i flere prekliniske sykdomsmodeller(F. eks.,) (Dai et al., 2016). I tillegg utgjør en økende forståelse av menneskelig variasjon nyere utfordringer for genterapi og driver innovasjon mot en sann personalisering av genredigering (Lessard et al., 2017; Scott Og Zhang, 2017). Nye teknologier som vSLENDR, ET aav-virus og CRISPR/Cas9-mediert teknologi for å erstatte defekte gener i nevroner og andre nervesystemceller, presser genredigeringsteknologier til nye grenser (Nishiyama et al., 2017).
Konklusjon
Infantil Neuroaksonal Dystrofi er en alvorlig nevrodegenerativ sykdom med en viss sykelighet og dødelighet. Denne sjeldne sykdommen gir en spennende mulighet til å revalidere tilgjengelige moduser av neste generasjons terapi og generere nyere. Emerging kongruens på de kliniske diagnostiske kriteriene FOR INAD forventes å gi en drivkraft mot forbedret molekylær diagnostikk. Denne fremgangen forventes å føre oss til å utvikle rimelige terapier som vil gi målbar forbedring I LIVSKVALITETEN TIL INAD-pasienter og forsinke eller forbedre sykdomsprogresjonen. Vi har fremhevet noen suksesshistorier i Batens sykdom og mukopolysakkaridosene som gir oss inspirasjonen til å stille de riktige spørsmålene for Å gjøre INAD-terapi til virkelighet. I tillegg bør voksende virale og ikke-virale tilnærminger for CRISPR / Cas9 – basert genredigering også åpne nyere terapeutiske veier for INAD.
Forfatterbidrag
PB og FA utarbeidet foreløpig utkast. SR og AC redigerte og la til flere seksjoner i manuskriptet. DF, AP og LP redigerte manuskriptet.
Interessekonflikt
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., Og Dennis, E. A. (1995). Hemming av kalsiumuavhengig fosfolipase A2 hindrer arakidonsyre inkorporering og fosfolipid remodeling I P388D1 makrofager. Proc. Natl. Acad. Sci. U. S. A. 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Effektiv CNS-målretting hos voksne mus ved intratekal infusjon av enkeltstrenget AAV9-GFP for genterapi av nevrologiske lidelser. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 for in vivo genterapi: løfte og hindringer. Mol. Ther. Nukleinsyrer 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Engel, L. A., Jing, Z., O ‘ Brien, D. E., Sun, M., Og Kotzbauer, Pt (2010). Katalytisk FUNKSJON AV PLA2G6 er svekket av mutasjoner assosiert med infantil neuroaxonal dystrofi, men ikke dystoni-parkinsonisme. PLoS En 5: e12897. doi: 10.1371 / tidsskrift.pone.0012897
PubMed Abstrakt | CrossRef Fulltekst
Gregory, A., Kurian, M. a., Maher, E. R., Hogarth, P., Og Hayflick, S. J. (1993). I PLA2G6-Assosiert Nevrodegenerasjon, eds M. P. Adam, H. H. Ardinger, Og R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregor, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). CNS-involvering I Fabrys sykdom: kliniske og avbildningsstudier før og etter 12 måneders enzymutskiftningsterapi. J. Arve. Metab. Dis. 27, 229–240. doi: 10.1023 / B: BOLI.0000028794.04349.91
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
A. S., A., A., A., a., a., a., a., a., a., a., a., a., a., a., a., a., a., a., et al. (2016). Genetisk analyse AV PLA2G6 i 22 indiske familier med infantil neuroaxonal dystrofi, atypisk sen-onset neuroaxonal dystrofi og dystoni parkinsonisme kompleks. PLoS En 11: e0155605. doi: 10.1371 / tidsskrift.pone.0155605
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
LaFountaine, Js, Fathe, K., Og Smyth, Hd (2015). Levering Og terapeutiske anvendelser av genredigeringsteknologier ZFNs. TALENs OG CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
A., A., A., A., A., A., A., A., A., A., A., A., a., a., a., a., a., a., a., a., a., a., a., a., a., a., a., a., a., a., et al. (2016). Analyse av proteinkodende genetisk variasjon hos 60 706 mennesker. Natur 536, 285-291. doi: 10.1038 / nature19057
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Menneskelig genetisk variasjon endrer CRISPR-Cas9 on-og off-targeting spesifisitet ved terapeutisk implisert loci. Proc. Natl. Acad. Sci. U. S. A. 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Levi, S., Og Finazzi, D. (2014). Neurodegenerasjon med hjernejernakkumulering: oppdatering på patogene mekanismer. Front. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Fulltekst | Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Mitokondriell lokalisering av cyklooksygenase-2 og kalsium-uavhengig fosfolipase A2 i humane kreftceller: implikasjon i apoptoseresistens. Exp. Celle Res. 306, 75-84. doi: 10.1016 / j.yexcr.2005.01.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Macauley, S. L. Og Sands, M. S. (2009). Lovende cns-rettet enzym erstatningsterapi for lysosomale lagrings sykdommer. Exp. Neurol. 218, 5–8. doi: 10.1016 / j.expneurol.2009.03.040
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Mehnaaz, L. (2016). Neurodegenerasjon med hjernejernakkumulering (NBIA) tidligere Hallervorden-Spatz sykdom. J. Assoc. Phys. India 64:132.
Google Scholar
Morton, J. E. V., Morton, J. E. V., Gregory, A., Gissen, P., Sonek, S., Et al. (2006). PLA2G6, som koder for en fosfolipase A2, er mutert i nevrodegenerative lidelser med høyt hjernejern. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Murphy, M. P. (2009). Hvordan mitokondrier produserer reaktive oksygenarter Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitã, C., Og Faria, A. (2016). Neurodegenerasjon med hjernejernakkumulering: en saksrapport. Dement. Neuropsykol. 10, 160–164. doi: 10.1590 / S1980-5764-2016DN1002014
PubMed Abstrakt | Kryssref Full Tekst | Google Scholar
Nishiyama, J., Mikuni, T. ,Og Yasuda, R. (2017). Virusmediert genomredigering via homologirettet reparasjon i mitotiske og postmitotiske celler i pattedyrhjernen. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j.neuron.2017.10.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., Og Farooqui, A. a. (2005). Distribusjon av kalsiumuavhengig fosfolipase A2 (iPLA2) i apehjerne. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed Abstrakt / Fulltekst / Google Scholar
Pardridge, W. M. (2002). Narkotika og gen rettet mot hjernen med molekylære Trojanske hester. Nat. Rev. Narkotika Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Pardridge, W. M. (2005a). Molekylærbiologi av blod-hjernebarrieren. Mol. Bioteknologi. 30, 57–70. doi: 10.1385 / MB: 30: 1:057
CrossRef Fulltekst | Google Scholar
Pardridge, W. M. (2005b). Blod-hjernebarrieren: flaskehals i hjernens medisinutvikling. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Kalsium-uavhengige fosfolipaser A2 og deres roller i biologiske prosesser og sykdommer. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., S Hryvnchez, V., S Hryvnchez, X., et al. (2017). Sykdomskorreksjon ved AAV-mediert genterapi i en ny musemodell av mukopolysakkaridose TYPE IIID. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg | ddx058
PubMed Abstrakt / CrossRef Fulltekst / Google Scholar
Scott, D. A., Og Zhang, F. (2017). Implikasjoner av menneskelig genetisk variasjon i CRISPR-basert terapeutisk genomredigering. Nat. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Zhao, K., Zhao, C., Zhang, X. H. Song, k. Og Ma, Z. A. (2006). Kalsium-uavhengig fosfolipase A2 lokaliserer og beskytter mitokondrier under apoptotisk induksjon av staurosporin. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Smith, A. J., Bainbridge, J. W. og Ali, R. R. (2012). Gent supplementation therapy for recessive former av arvelige retinal dystrophies. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Sorrentino, N. C., D ‘ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). En svært utskilt sulfamidase konstruert for å krysse blod-hjernebarrieren korrigerer hjernelesjoner av mus med mukopolysakkaridoser type iiia. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Sorrentino, N. C., Og Fraldi, A. (2016). Hjernemålretting i MPS-IIIA. Pediatr. Endokrinol. Åp 13 (Suppl. 1), 630–638.
Google Scholar
J., W., Wolfman, N., Shaffer, M. Seehra, J. Og Jones, S. S. (1997). En ny cytosolisk kalsium-uavhengig fosfolipase A2 inneholder åtte ankyrinmotiver. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
U. S. Food And Drug Administration (2017). FDA Godkjenner Første Behandling for En Form For Batten Sykdom. Tilgjengelig på: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., Og Gottlieb, R. A. (2002). Hemming av mitokondriell kalsiumuavhengig fosfolipase A2 (iPLA2) demper mitokondrielt fosfolipidtap og er hjertebeskyttende. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
Yu-Wai-Man, P. (2016). Genetisk manipulasjon for arvelige neurodegenerative sykdommer: myte eller virkelighet? Br. J. Oftalmol. 100:1322. doi: 10.1136 / bjoftalmol-2015-308329
PubMed Abstrakt / Fulltekst / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et al. (2010). Beskyttelse av pankreas beta-celler etter gruppe via fosfolipase A (2) – mediert reparasjon av mitokondriell membranperoksydasjon. Endokrinologi 151, 3038-3048. doi: 10.1210 / en.2010-0016
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar