Introduzione
La distrofia neuroassonale infantile (INAD) è una malattia neurodegenerativa rara autosomica recessiva di frequenza sconosciuta. L’insorgenza dei sintomi si verifica generalmente tra i 6 mesi e i 3 anni di età. Prima di quel momento, i bambini si sviluppano normalmente. Il primo sintomo di INAD può essere il rallentamento del raggiungimento di normali pietre miliari dello sviluppo o la regressione nelle pietre miliari dello sviluppo (Ramanadham et al., 2015). L’ipotonia del tronco, lo strabismo e il nistagmo sono i primi sintomi della malattia (Gregory et al., 2017). La progressione della malattia è rapida e man mano che progredisce, si perdono più abilità acquisite. I muscoli diventano presto ipotonici e in seguito diventano spastici (Levi e Finazzi, 2014). Alla fine, tutto il controllo muscolare volontario è perso. La debolezza muscolare può anche portare a difficoltà nell’alimentazione e nella respirazione. Oltre al nistagmo, alcuni bambini sperimentano la perdita della vista. Le funzioni cognitive vengono gradualmente perse e si sviluppa la demenza. La durata della vita è generalmente 5 a 10 anni (Gregory et al., 1993; Jardim et al., 2004; Macauley e Sands, 2009).
Patologia molecolare
La distrofia neuroassonale infantile appartiene a una famiglia di disturbi neurodegenerativi che include la distrofia neuroassonale atipica a esordio tardivo (ANAD) e il complesso di parkinsonismo distonico (DPC). La maggior parte dei casi di INAD sono associati a mutazioni eterozigoti omozigoti o composti nel gene PLA2G6 che influenzano l’attività catalitica del suo prodotto proteico (Engel et al., 2010). Il gene PLA2G6 codifica un gruppo tramite la proteina fosfolipasi A2 indipendente dal calcio (PLA2G6 o iPLA2ß, ∼85/88 kDa) con una lipasi e sette domini contenenti ripetizioni di ankyrin (Tang et al., 1997). PLA2G6 idrolizza la catena acilica sn-2 dei fosfolipidi, generando acidi grassi liberi e lisofosfolipidi.
I fosfolipidi nella membrana interna dei mitocondri sono ricchi di acidi grassi insaturi nella posizione sn-2, in particolare cardiolipina (Seleznev et al., 2006). Questi acidi grassi insaturi sono particolarmente vulnerabili alle abbondanti specie reattive dell’ossigeno prodotte dai mitocondri (Murphy, 2009) con conseguente fosfolipidi perossidati nella membrana interna dei mitocondri. PLA2G6 si localizza ai mitocondri (Williams e Gottlieb, 2002; Liou et al., 2005) coerente con un aumento della domanda di idrolisi degli acidi grassi perossidati nella posizione sn – 2 dei fosfolipidi che porta a fosfolipidi rimodellati (Balsinde et al., 1995; Zhao et al., 2010). Quando PLA2G6 è difettoso, l’integrità della membrana interna dei mitocondri è danneggiata. PLA2G6 localizza anche l’assone (Ong et al., 2005; Seleznev et al., 2006) indicando una domanda localizzata aumentata di fosfolipidi che ritocca là pure. La manifestazione di tale accumulo nel cervello è unica per le aree cerebrali chiave, come i gangli della base che hanno portato a vari nomi per la stessa patogenesi molecolare sottostante che coinvolge PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
L’analisi ultrastrutturale dei neuroni nei topi knockout PLA2G6 è coerente con questa patologia molecolare. Sono stati osservati mitocondri con creste ramificate e tubolari, mitocondri con creste degenerate, assoni con collasso del citoscheletro e perdita parziale della membrana ai terminali degli assoni (Beck et al., 2011). A livello microscopico, queste caratteristiche appaiono come gonfiori assonali e corpi sferoidi nei terminali pre-sinaptici (Figura 1) nel sistema nervoso centrale o periferico.
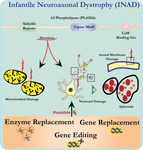
Figura 1. La distrofia neuroassonale infantile (INAD) è una malattia neurodegenerativa correlata a mutazioni nel gene PLA2G6. Varie mutazioni di PLA2G6 portano alla fosfolipasi A2 disfunzionale che porta a difetti della membrana mitocondriale e assonale. Questi difetti causano danni neuronali visualizzati come gonfiori assonali e accumulo di sferoidi pre-sinaptici. Sostituzione enzimatica per ripristinare le funzioni, sostituzione genica o modifica per correggere il PLA2G6 difettoso sono proposte strategie terapeutiche.
Diagnostica Molecolare e delle Malattie Rare di Responsabilizzazione del Paziente
a parte le specifiche clinici, elettrofisiologici e funzionalità di imaging, prima la disponibilità di next generation sequencing, biopsie cutanee, mostrando assonale gonfiori e corpi sferoidali in pre-terminali sinaptici nella parte centrale o il sistema nervoso periferico sono stati i criteri diagnostici per la conferma dell’INAD (Gregory et al., 2017; Iodice et al., 2017). Spesso, sono state necessarie più biopsie per confermare la diagnosi. Le famiglie generalmente aspettavano molti anni per una diagnosi. Con il costo decrescente del sequenziamento genico e genomico, la disponibilità di test genetici mirati con laboratori diagnostici per malattie neurologiche non diagnosticate e la crescente consapevolezza dei medici della disponibilità di diagnostica genetica, le famiglie ricevono la diagnosi più rapidamente; a volte entro un anno dalla comparsa del primo sintomo. I bambini in queste famiglie sono ancora giovani e le famiglie sono motivate a collaborare con gli scienziati per trovare un trattamento per la malattia dei loro figli. Al fine di finanziare il processo di ricerca, un gruppo di genitori di pazienti INAD ha formato la Fondazione INADcure. La fondazione ha raccolto ingenti fondi per la ricerca e sta collaborando con Rare Genomics Institute per guidarli nell’assegnazione di borse di ricerca. È interessante notare che un’analisi genetica 2016 di 22 famiglie indiane con INAD, ANAD e DPC ha rilevato che le famiglie 10 / 22 (45.45%) mancavano di mutazioni nella regione di codifica del gene PLA2G6 (Kapoor et al., 2016). La mancata identificazione di mutazioni deleterie nella regione codificante di PLA2G6 evidenzia che i futuri sforzi diagnostici molecolari richiederebbero un sequenziamento genico completo per identificare mutazioni nelle regioni introniche e regolatorie del gene PLA2G6 in pazienti affetti da INAD. Inoltre, i casi INAD in cui non si osservano mutazioni associate a PLA2G6 suggeriscono che la causa della malattia potrebbe essere dovuta a mutazioni in geni diversi da PLA2G, che devono essere esplorati.
Possibili terapie
La maggior parte delle terapie prese in considerazione per una malattia rara come l’INAD che coinvolge un enzima difettoso sono la sostituzione enzimatica, la sostituzione genica o la correzione genica. Quando una carenza enzimatica è causata da un difetto genetico recessivo, si presume che la sostituzione enzimatica o la supplementazione possano correggere il problema (Smith et al., 2012; Yu-Wai-Man, 2016). Tuttavia, gli esperimenti per dimostrare che le terapie che forniscono il gene o l’enzima corretto salveranno il fenotipo INAD devono ancora essere eseguiti e testati.
Terapia enzimatica sostitutiva (ERT)
Poiché il cervello è l’organo primario interessato in INAD, la terapia enzimatica sostitutiva per INAD richiederebbe molto probabilmente l’infusione dell’enzima nel cervello. Nel 2017, Biomarin ha ricevuto l’approvazione della FDA per tripeptidil peptidasi 1 (cerliponasi alfa) come trattamento per la causa sottostante della malattia di Batten, la carenza di tripeptidil peptidasi (TPP1) un enzima lisosomiale (US Food Drug Administration, 2017). Tripeptidil peptidasi 1 (cerliponasi alfa; ∼59 kDa) è il primo ERT ad essere somministrato direttamente nel liquido cerebrospinale (CSF) del cervello. Altri farmaci ERT che possono essere somministrati nel CSF sono in studi clinici (Jardim et al., 2004; Macauley e Sands, 2009).
L’efficacia della tripeptidil peptidasi 1 (cerliponasi alfa) sulla capacità di deambulazione dei pazienti con malattia di Batten dimostra che la terapia enzimatica sostitutiva per le malattie che colpiscono il cervello è teoricamente possibile. Il targeting dell’enzima sostitutivo nei mitocondri per l’INAD sarà più impegnativo del targeting lisosomiale che aveva già avuto successo da IV ERT per un lungo periodo, come nel caso della malattia di Gaucher. Quindi, ci sono ancora molti problemi biologici specifici per INAD che devono essere risolti:
• È sufficiente o anche PLA2G6 dovrà essere localizzato sull’assone e, in caso affermativo, come?
• C’è una particolare necessità di affrontare il targeting del sistema nervoso periferico?
• Un PLA2G6 parziale può salvare la sua carenza o è necessaria una proteina intera?
• Se l’enzima PLA2G6 non è puro al 100%, quali sono i suoi contaminanti e sono sicuri?
Un ulteriore problema è che la terapia enzimatica sostitutiva richiede il posizionamento iniziale di un catetere intracerebroventricolare e frequenti infusioni. Il posizionamento del catetere richiede anestesia e le infusioni possono richiedere anestesia a seconda della cooperazione dei pazienti. Affrontare le preoccupazioni dei neurologi pediatrici e dei genitori di bambini con INAD per quanto riguarda l’uso dell’anestesia su pazienti INAD con informazioni appropriate richiederà un ulteriore sforzo.
Terapia genica / Sostituzione genica
Il gene PLA2G umano è di circa 70 kb con 17 esoni e 2 esoni alternativi. La trascrizione più lunga di codifica delle proteine, tuttavia, è solo 3,3 kb e la sequenza di codifica delle proteine è poco più di 2,4 kb, che può essere facilmente confezionata in un carico vettoriale virale. Per prima cosa esploreremo un altro disturbo da accumulo lisosomiale nel SNC che ha dati preclinici da confrontare con l’INAD. La mancanza di dati preclinici di ERT in INAD è un significativo divario di conoscenza nel campo, motivo per cui è fondamentale osservare altri modelli preclinici di disturbi del SNC che sono sottoposti a studi di terapia genica. In precedenza, uno studio preclinico sui topi ha affrontato la terapia genica per il disturbo da accumulo lisosomiale mucopolisaccaridosi di tipo IIIA (MPS-IIIA) confezionando la versione corretta del gene sulfamidasi all’interno di un vettore virale (Sorrentino et al., 2013). Lo studio ha approfittato della crescente comprensione della barriera emato-encefalica (BBB) che regola attivamente il trasporto di grandi molecole dal sangue al SNC mediante un processo chiamato transcitosi (Pardridge, 2005b; Sorrentino e Fraldi, 2016).
La transcitosi comporta l’endocitosi dei ligandi sul lato luminale, mediata da recettori specifici del ligando (ad esempio, recettore dell’insulina, recettore della transferrina e recettore delle lipoproteine a bassa densità, ecc.,) arricchito sull’endotelio capillare (Pardridge, 2005a). Questo è seguito dal movimento del carico endocytosed attraverso il citoplasma dell’endotelio e infine dall’esocitosi sul lato abluminale (cervello), consegnando così efficacemente il carico attraverso il BBB (Pardridge, 2002). Lo studio preclinico MPS-IIIA ha prodotto una proteina chimerica sulfamidasi che conteneva un dominio BBB-binding da apolipoproteina B per facilitare l’assorbimento da parte dell’endotelio e anche un peptide segnale da iduronato-2-sulfatasi per aiutare un’esocitosi efficiente verso il lato abluminale del BBB (Sorrentino et al., 2013). Un carico vettoriale virale che codifica una sulfamidasi chimerica è stato quindi caricato su un sierotipo 2/8 del virus adeno-associato (AAV) mirato al fegato (Sorrentino e Fraldi, 2016). Così il fegato ha servito come una fabbrica interna che ha fornito un rifornimento costante della chimerico-sulfamidasi, che ha provocato un aumento di 10-15% nell’attività della sulfamidasi del cervello anche 7 mesi dopo la terapia genica del fegato. Questo aumento dei livelli di attività della sulfamidasi cerebrale ha portato a un miglioramento quantificabile nella patologia cerebrale e nei risultati comportamentali nel modello murino di MPS-IIIA (Sorrentino et al., 2013).
Se uno studio simile è condotto per INAD, risponderebbe a diverse domande critiche sulla sostituzione enzimatica per la terapia INAD. In alternativa, la somministrazione intra-vascolare o intra-CSF di prodotti di terapia genica a base di AAV9 può colpire direttamente il SNC (Bey et al., 2017; Roca et al., 2017). Tuttavia, le mutazioni multiple che i pazienti di INAD hanno sul loro gene PLAG26 fornisce le sfide uniche alla terapia genica. Sebbene la dimensione del gene PLA2G6 di 2-3 kb non dovrebbe rappresentare un problema per il suo inserimento in un vettore virale, tuttavia, le complicazioni normative derivanti dalla correzione di PLA2G6 sono difficili da prevedere. Le complicazioni regolatorie diventano incontrollabili specialmente se la terapia genica non può essere localizzata correttamente all’interno dei tessuti bersaglio. La terapia enzimatica sostitutiva può anche essere un problema se alcuni prodotti mutanti risultano essere negativi dominanti al PLAG26 wild-type.
La maggior parte delle sfide delle possibili terapie per l’INAD deriva dal fatto che si tratta di una malattia ultra-rara. Tuttavia, il fatto che l’INAD abbia un’eziologia genetica nota fornisce strade per possibili terapie. Inoltre, qualsiasi terapia efficace per INAD riceverà lo status di farmaco orfano e tutta la protezione che ottiene perché INAD è una malattia rara. Pertanto, nonostante tutte le sfide di cui sopra, lo status di farmaco orfano fornisce un forte incentivo per i ricercatori di malattie rare e l’industria biotecnologica, per non parlare di quando la causa genetica è già nota.
Gene/Base Editing
A partire dal 2017, sono state osservate almeno 277 mutazioni missense nel gene PLA2G6 umano (Lek et al., 2016). Solo una piccola percentuale di mutazioni PLA2G6 include spostamenti di frame, indel, mutazioni senza senso e mutazioni nei siti di giunzione (Morgan et al., 2006). Quindi correggere il gene nella popolazione di cellule bersaglio o correggere le basi del DNA mutato è un’attraente via terapeutica. Nel corso degli anni, sono stati sviluppati diversi strumenti per l’editing genetico e l’editing genomico basato su CRISPR/Cas9 viene salutato come una tecnologia innovativa con uno studio clinico pianificato nel 2018. Queste tecnologie utilizzano una proteina legante il DNA che può anche fendere il filamento in un modo specificato per fare spazio per l’inserimento di una nuova sequenza di DNA o la correzione della base deleteria del DNA (LaFountaine et al., 2015). La tecnologia CRISPR / Cas9 ha già prodotto risultati terapeutici sorprendenti in diversi modelli di malattie precliniche (ad esempio, distrofia muscolare di Duchene, malattie metaboliche epatiche, ecc.,) (Dai et al., 2016). Inoltre, una crescente comprensione della variazione umana sta ponendo nuove sfide alla terapia genica e guidando l’innovazione verso una vera personalizzazione dell’editing genico (Lessard et al., 2017; Scott e Zhang, 2017). Nuove tecnologie come vSLENDR, un virus AAV e la tecnologia mediata da CRISPR / Cas9 per sostituire i geni difettosi nei neuroni e in altre cellule del sistema nervoso stanno spingendo le tecnologie di editing genetico verso nuove frontiere (Nishiyama et al., 2017).
Conclusione
La distrofia neuroassonale infantile è una grave malattia neurodegenerativa con una certa morbilità e mortalità. Questa malattia rara offre un’entusiasmante opportunità di rivalutare le modalità disponibili di terapia di nuova generazione e di generarne di nuove. La congruenza emergente sui criteri diagnostici clinici per INAD dovrebbe fornire un impulso verso la diagnostica molecolare migliorata. Si prevede che questo progresso ci porterà a sviluppare terapie a prezzi accessibili che fornirebbero un miglioramento quantificabile della qualità della vita dei pazienti INAD e ritarderebbero o migliorerebbero la progressione della malattia. Abbiamo evidenziato alcune storie di successo nella malattia di Batten e le mucopolisaccaridosi che ci forniscono l’ispirazione per porre le domande giuste per rendere la terapia INAD una realtà. Inoltre, la crescita di approcci virali e non virali per l’editing genetico basato su CRISPR/Cas9 dovrebbe anche aprire nuove vie terapeutiche per l’INAD.
Contributi dell’autore
PB e FA hanno preparato il progetto preliminare. SR e AC hanno modificato e aggiunto altre sezioni al manoscritto. DF, AP e LP hanno curato il manoscritto.
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., e Dennis, E. A. (1995). L’inibizione della fosfolipasi A2 indipendente dal calcio impedisce l’incorporazione dell’acido arachidonico e il rimodellamento del fosfolipide nei macrofagi P388D1. Proc. Natl. Acad. Sic. USA 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Efficiente targeting del SNC in topi adulti mediante infusione intratecale di AAV9-GFP a singolo filamento per la terapia genica di disturbi neurologici. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstract / CrossRef Testo completo / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 per la terapia genica in vivo: promessa e ostacoli. Mol. Ther. Acidi nucleici 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstract / CrossRef Testo completo / Google Scholar
Engel, L. A., Jing, Z., O’Brien, D. E., Sun, M., e Kotzbauer, PT (2010). La funzione catalitica di PLA2G6 è compromessa da mutazioni associate alla distrofia neuroassonale infantile ma non al distonia-parkinsonismo. PLoS Uno 5: e12897. doi: 10.1371 / giornale.pone.0012897
PubMed Abstract | CrossRef Testo completo
Gregory, A., Kurian, Maher, E. R., Hogarth, P., e Hayflick, S. J. (1993). in PLA2G6-associato Neurodegenerazione, eds MP Adam, HH Ardinger, e RA Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Coinvolgimento del SNC nella malattia di Fabry: studi clinici e di imaging prima e dopo 12 mesi di terapia enzimatica sostitutiva. J. Eredita. Metab. Dis. 27, 229–240. doi: 10.1023 / B: BOLI.0000028794.04349.91
PubMed Abstract / CrossRef Full Text / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Piuttosto, M. I., Bhat, V., Gopinath, S., et al. (2016). Analisi genetica di PLA2G6 in 22 famiglie indiane con distrofia neuroassonale infantile, distrofia neuroassonale atipica a esordio tardivo e complesso di parkinsonismo distonico. PLoS Uno 11:e0155605. doi: 10.1371 / giornale.pone.0155605
PubMed Abstract | CrossRef Testo completo / Google Scholar
LaFountaine, J. S., Fathe, K., e Smyth, HD (2015). Delivery and therapeutic applications of gene editing technologies ZFNs. TALENs, e CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed Abstract | CrossRef Testo completo / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analisi della variazione genetica proteina-codificante in 60.706 esseri umani. Natura 536, 285-291. doi: 10.1038/nature19057
PubMed Astratto | CrossRef Testo Completo | Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T. MacArthur, D. G., et al. (2017). Variazione genetica umana altera CRISPR-Cas9 on-e off-targeting specificità a loci terapeuticamente implicati. Proc. Natl. Acad. Sic. U. S. A. 14, E11257–E11266. doi: 10.1073 / pnas.1714640114
PubMed Abstract | CrossRef Testo completo / Google Scholar
Levi, S., e Finazzi, D. (2014). Neurodegenerazione con accumulo di ferro cerebrale: aggiornamento sui meccanismi patogeni. Anteriore. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Testo Completo | Google Scholar
Liu, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Localizzazione mitocondriale della cicloossigenasi-2 e della fosfolipasi A2 calcio-indipendente nelle cellule tumorali umane: implicazione nella resistenza all’apoptosi. Scad. Risoluzione 306, 75-84. doi: 10.1016 / j. yexcr.2005.01.011
PubMed Abstract | CrossRef Full Text / Google Scholar
Macauley, SL, e Sands, MS (2009). Promettente terapia enzimatica sostitutiva diretta al SNC per le malattie da accumulo lisosomiale. Scad. Neurolo. 218, 5–8. doi: 10.1016 / j.expneurol.2009.03.040
PubMed Abstract | CrossRef Testo completo / Google Scholar
Mehnaaz, L. (2016). Neurodegenerazione con accumulo di ferro cerebrale (NBIA) precedentemente malattia di Hallervorden-Spatz. J. Assoc. Phys. India 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K., Morton, J. E. V., di Gregorio, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, che codifica per una fosfolipasi A2, è mutato nei disturbi neurodegenerativi con ferro cerebrale alto. NAT. Genet. 38, 752–754. doi: 10.1038 | ng1826
PubMed Abstract / CrossRef Testo completo / Google Scholar
Murphy, MP (2009). Come i mitocondri producono specie reattive dell’ossigeno. Biochimica. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstract / CrossRef Full Text / Google Scholar
Nel 2016 si trasferisce in Francia, dove si trasferisce in Italia. Neurodegenerazione con accumulo di ferro cerebrale:un caso clinico. Dement. Neuropsicolo. 10, 160–164. doi: 10.1590 / S1980-5764-2016DN1002014
PubMed Abstract / CrossRef Testo completo / Google Scholar
Nishiyama, J., Mikuni, T. e Yasuda, R. (2017). Modifica del genoma mediata dal virus tramite riparazione diretta dall’omologia in cellule mitotiche e postmitotiche nel cervello dei mammiferi. Neurone 96, 755.e5-768.e5. doi: 10.1016 / j.neurone.2017.10.004
PubMed Abstract | CrossRef Testo completo / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., e Farooqui, A. A. (2005). Distribuzione della fosfolipasi A2 calcio-indipendente (iPLA2) nel cervello della scimmia. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed Abstract / CrossRef Testo completo / Google Scholar
Pardridge, W. M. (2002). Drug and gene targeting per il cervello con cavalli di Troia molecolari. NAT. Rev. Droga Discov. 1, 131–139. doi: 10.1038 | nrd725
PubMed Abstract / CrossRef Full Text / Google Scholar
Pardridge, W. M. (2005a). Biologia molecolare della barriera emato-encefalica. Mol. Biotecnologia. 30, 57–70. doi: 10.1385 / MB:30: 1:057
CrossRef Testo completo / Google Scholar
Pardridge, W. M. (2005b). La barriera emato-encefalica: collo di bottiglia nello sviluppo di farmaci cerebrali. NeuroRx 2, 3-14.
Google Scholar
Il sito utilizza cookie tecnici e cookie di profilazione di terze parti. (2015). Fosfolipasi calcio-indipendenti A2 e loro ruolo nei processi biologici e nelle malattie. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstract / CrossRef Testo completo / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Correzione della malattia mediante terapia genica mediata da AAV in un nuovo modello murino di mucopolisaccaridosi di tipo IIID. Ronzio. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg / ddx058
PubMed Abstract / CrossRef Full Text / Google Scholar
Scott, D. A., e Zhang, F. (2017). Implicazioni della variazione genetica umana nell’editing terapeutico del genoma basato su CRISPR. NAT. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstract / CrossRef Testo completo / Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Canzone, K., e Ma, Z. A. (2006). La fosfolipasi A2 calcio-indipendente localizza e protegge i mitocondri durante l’induzione apoptotica da parte della staurosporina. J. Biol. Chimica. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstract | CrossRef Testo completo / Google Scholar
Nel 2012 è stato pubblicato il primo album in studio del gruppo. Terapia di integrazione genica per forme recessive di distrofie retiniche ereditarie. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstract / CrossRef Testo completo / Google Scholar
Sorrentino, N. C., D’Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Una sulfamidasi altamente secreta progettata per attraversare la barriera emato-encefalica corregge le lesioni cerebrali dei topi con mucopolisaccaridosi di tipo IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed Abstract / CrossRef Testo completo / Google Scholar
Sorrentino, N. C., e Fraldi, A. (2016). Brain targeting in MPS-IIIA. Pediatria. Endocrinolo. Apoc.13(Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., e Jones, S. S. (1997). Una nuova fosfolipasi citosolica indipendente dal calcio A2 contiene otto motivi di ankyrin. J. Biol. Chimica. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstract | CrossRef Testo completo / Google Scholar
U. S. Food and Drug Administration (2017). La FDA approva il primo trattamento per una forma di malattia di Batten. Disponibile presso: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., e Gottlieb, R. A. (2002). L’inibizione della fosfolipasi mitocondriale indipendente dal calcio A2 (iPLA2) attenua la perdita di fosfolipidi mitocondriali ed è cardioprotettiva. Biochimica. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstract / CrossRef Full Text / Google Scholar
Yu-Wai-Man, P. (2016). Manipolazione genetica per malattie neurodegenerative ereditarie: mito o realtà? Fr. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed Abstract / CrossRef Testo completo / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., et al. (2010). Protezione delle beta-cellule pancreatiche per gruppo TRAMITE la riparazione mediata dalla fosfolipasi A(2) della perossidazione della membrana mitocondriale. Endocrinologia 151, 3038-3048. doi: 10.1210 / it.2010-0016
PubMed Abstract / CrossRef Full Text / Google Scholar