Introducere
distrofia Neuroaxonală infantilă (INAD) este o boală neurodegenerativă rară autosomală recesivă cu frecvență necunoscută. Debutul simptomelor apare în general între 6 luni și 3 ani. Înainte de acest timp, sugarii se dezvoltă normal. Primul simptom al INAD poate fi încetinirea atingerii etapelor normale de dezvoltare sau regresia în etapele de dezvoltare (Ramanadham și colab., 2015). Hipotonia trunchiului, strabismul și nistagmusul sunt simptome precoce ale bolii (Gregory și colab., 2017). Progresia bolii este rapidă și, pe măsură ce progresează, se pierd mai multe abilități dobândite. Mușchii devin în curând hipotonici și mai târziu devin spastici (Levi și Finazzi, 2014). În cele din urmă, se pierde tot controlul voluntar al mușchilor. Slăbiciunea musculară poate duce, de asemenea, la dificultăți în hrănire și respirație. În plus față de nistagmus, unii copii suferă de pierderea vederii. Funcțiile Cognitive se pierd treptat și se dezvoltă demența. Durata de viață este în general de 5 până la 10 ani (Gregory și colab., 1993; Jardim și colab., 2004; Macauley și Sands, 2009).
Patologie Moleculară
distrofia Neuroaxonală infantilă aparține unei familii de tulburări neurodegenerative care include distrofia neuroaxonală atipică cu debut tardiv (ANAD) și complexul de Parkinsonism distonie (DPC). Majoritatea cazurilor de INAD sunt asociate cu mutații heterozigote homozigote sau compuse în gena PLA2G6 care afectează activitatea catalitică a produsului său proteic (Engel și colab., 2010). Gena PLA2G6 codifică un grup prin proteina A2 A fosfolipazei independente de calciu (PLA2G6 sau ipla2 Irak, 85/88 kDa) cu o lipază și șapte domenii care conțin repetare ankirină (Tang și colab., 1997). PLA2G6 hidrolizează lanțul acil SN-2 al fosfolipidelor, generând acizi grași liberi și lizofosfolipide.
fosfolipidele din membrana interioară a mitocondriilor sunt bogate în acizi grași nesaturați în poziția sn-2, în special cardiolipina (Seleznev și colab., 2006). Acești acizi grași nesaturați sunt deosebit de vulnerabili la speciile abundente de oxigen reactiv produse de mitocondrii (Murphy, 2009), rezultând fosfolipide peroxidate în membrana interioară a mitocondriilor. PLA2G6 se localizează în mitocondrii (Williams și Gottlieb, 2002; Liou și colab., 2005) în concordanță cu o cerere crescută de hidroliză a acizilor grași peroxidați în poziția SN-2 a fosfolipidelor care duce la fosfolipide remodelate (Balsinde și colab., 1995; Zhao și colab., 2010). Când PLA2G6 este defect, integritatea membranei interioare a mitocondriilor este deteriorată. PLA2G6 se localizează și la axon (Ong și colab., 2005; Seleznev și colab., 2006) indicând o cerere localizată crescută pentru remodelarea fosfolipidelor și acolo. Manifestarea unei astfel de acumulări în creier este unică pentru zonele cheie ale creierului, cum ar fi ganglionii bazali, care au dus la diferite nume pentru aceeași patogeneză moleculară care implică PLAG26 (Mehnaaz, 2016; Nassif și colab., 2016).
analiza ultrastructurii neuronilor la șoarecii knockout PLA2G6 este în concordanță cu această patologie moleculară. Mitocondriile cu cristae ramificate și tubulare, mitocondriile cu cristae degenerate, axonii cu colapsul citoscheletului și pierderea parțială a membranei la terminalele axonului au fost observate (Beck și colab., 2011). La nivel microscopic, aceste caracteristici apar ca umflături axonale și corpuri sferoide în terminalele pre-sinaptice (Figura 1) din sistemul nervos central sau periferic.
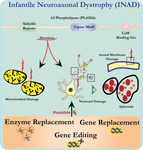
Figura 1. Distrofia neuroaxonală infantilă (INAD) este o tulburare neurodegenerativă legată de mutații ale genei PLA2G6. Diverse mutații ale PLA2G6 duc la fosfolipaza A2 disfuncțională care duce la defecte ale membranei mitocondriale și axonale. Aceste defecte provoacă leziuni neuronale vizualizate ca umflături axonale și acumularea de sferoizi pre-sinaptici. Înlocuirea enzimei pentru a restabili funcțiile, înlocuirea genelor sau editarea pentru a corecta pla2g6 defect sunt propuse strategii terapeutice.
diagnosticul Molecular și abilitarea pacienților cu boli Rare
în afară de caracteristicile clinice, electrofiziologice și imagistice specifice, înainte de disponibilitatea secvențierii de generație următoare, biopsiile cutanate care prezintă umflături axonale și corpuri sferoide în terminalele pre-sinaptice din sistemul nervos central sau periferic au fost criteriile de diagnostic pentru confirmarea INAD (Gregory și colab., 2017; Iodice și colab., 2017). Adesea, au fost necesare biopsii multiple pentru a confirma diagnosticul. În general, familiile au așteptat mulți ani pentru un diagnostic. Odată cu scăderea costului secvențierii genei și genomului, disponibilitatea testării panoului genetic vizat cu laboratoare de diagnostic pentru boli neurologice nediagnosticate și creșterea gradului de conștientizare a medicilor cu privire la disponibilitatea diagnosticului genetic, familiile primesc diagnosticul mai rapid; uneori în decurs de un an de la apariția primului simptom. Copiii din aceste familii sunt încă tineri, iar familiile sunt motivate să colaboreze cu oamenii de știință pentru a găsi un tratament pentru boala copiilor lor. Pentru a finanța procesul de cercetare, un grup de părinți ai pacienților INAD au format Fundația INADcure. Fundația a strâns fonduri substanțiale pentru cercetare și colaborează cu Institutul de Genomică rară pentru a-i îndruma în acordarea granturilor de cercetare. Interesant este că o analiză genetică din 2016 a 22 de familii indiene cu INAD, ANAD și DPC a constatat că 10/22 familii (45,45%) nu aveau mutații în regiunea de codificare a genei PLA2G6 (Kapoor și colab., 2016). Eșecul identificării mutațiilor dăunătoare în regiunea de codificare a PLA2G6 evidențiază faptul că eforturile viitoare de diagnostic molecular ar necesita secvențierea genelor întregi pentru a identifica mutațiile în regiunile intronice și de reglementare ale genei PLA2G6 la pacienții afectați de INAD. În plus, cazurile INAD în care mutațiile asociate PLA2G6 nu sunt observate sugerează că cauza bolii ar putea fi datorată mutației în alte gene decât PLA2G, care trebuie explorată.
terapii posibile
majoritatea terapiilor luate în considerare pentru o boală rară precum INAD care implică o enzimă defectă sunt înlocuirea enzimei, înlocuirea genelor sau corecția genelor. Atunci când o deficiență enzimatică este cauzată de un defect genetic recesiv, se presupune că înlocuirea sau suplimentarea enzimei poate corecta problema (Smith și colab., 2012; Yu-Wai-Man, 2016). Cu toate acestea, experimentele pentru a demonstra că terapiile care furnizează gena sau enzima corectă vor salva fenotipul INAD nu au fost încă efectuate și testate.
terapia de substituție enzimatică (ERT)
deoarece creierul este organul principal afectat în INAD, terapia de substituție enzimatică pentru INAD ar necesita cel mai probabil perfuzarea enzimei în creier. În 2017, Biomarin a primit aprobarea FDA pentru tripeptidil peptidaza 1 (cerliponaza alfa) ca tratament pentru cauza principală a bolii Batten, deficiența tripeptidil peptidazei (TPP1) o enzimă lizozomală (US Food Drug Administration, 2017). Tripeptidil peptidază 1 (cerliponază alfa; 59 kDa) este primul ERT administrat direct în lichidul cefalorahidian (LCR) al creierului. Alte medicamente ERT care pot fi administrate în LCR sunt în studii clinice (Jardim și colab., 2004; Macauley și Sands, 2009).
eficacitatea tripeptidil peptidazei 1 (cerliponaza alfa) asupra capacității de mers a pacienților cu boală Batten demonstrează că terapia de substituție enzimatică pentru bolile care afectează creierul este teoretic posibilă. Direcționarea enzimei de înlocuire în mitocondrii pentru INAD va fi mai dificilă decât direcționarea lizozomală, care a avut deja succes de ERT IV de mult timp, ca în cazul bolii Gaucher. Prin urmare, există încă multe probleme biologice specifice INAD care trebuie rezolvate:
• terapia pentru INAD are nevoie de mecanisme care pot transporta PLA2G6 în celulă?
• ce mecanism poate fi folosit pentru a localiza PLA2G6 la mitocondrii?
• este suficient sau va PLA2G6, de asemenea, trebuie să fie localizate la axon și dacă da, cum?
• există o nevoie specială de a aborda direcționarea sistemului nervos periferic?
• ce este un solvent adecvat pentru livrarea produsului genei PLA2G6 în LCR?
• dacă enzima PLA2G6 nu este 100% pură, care sunt contaminanții săi și sunt siguri?
• vor exista complicații intravasculare generalizate datorate perfuziei enzimei PLA2G6 în SNC? Are doza de perfuzie vreo relevanță pentru astfel de complicații?
o problemă suplimentară este că terapia de substituție enzimatică necesită plasarea inițială a unui cateter intracerebroventricular și perfuzii frecvente. Plasarea cateterului necesită anestezie, iar perfuziile pot necesita anestezie în funcție de cooperarea pacienților. Abordarea preocupărilor neurologilor pediatrici și ale părinților copiilor cu INAD în ceea ce privește utilizarea anesteziei la pacienții cu INAD cu informații adecvate va necesita eforturi suplimentare.
terapia genică/înlocuirea genelor
gena umană PLA2G este în jur de 70 kb cu 17 exoni și 2 exoni alternativi. Cu toate acestea, cea mai lungă transcriere a codării proteinelor este de doar 3,3 kb, iar secvența de codare a proteinelor este puțin peste 2,4 kb, care poate fi ușor ambalată într-o încărcătură vectorială virală. Vom explora mai întâi o altă tulburare de stocare lizozomală în SNC care are Date preclinice pentru a fi apoi comparate cu INAD. Lipsa datelor preclinice ale ERT în INAD este un decalaj semnificativ de cunoștințe în domeniu, motiv pentru care este crucial să observăm alte modele preclinice ale tulburărilor SNC care sunt supuse studiilor de terapie genică. Anterior, un studiu preclinic la șoareci a abordat terapia genică pentru tulburarea de stocare lizozomală mucopolizaharidoză de tip IIIA (MPS-IIIA) prin ambalarea versiunii corecte a genei sulfamidazei în interiorul unui vector viral (Sorrentino și colab., 2013). Studiul a profitat de înțelegerea tot mai mare a barierei hematoencefalice (BBB), care reglementează în mod activ transportul moleculelor mari din sânge în SNC printr-un proces numit transcitoză (Pardridge, 2005b; Sorrentino și Fraldi, 2016).
Transcitoza implică endocitoza liganzilor pe partea luminală, mediată de receptorii specifici ligandului (de exemplu, receptorul insulinei, receptorul transferinei și receptorul lipoproteinelor cu densitate mică etc.,) îmbogățit pe endoteliul capilar (Pardridge, 2005A). Aceasta este urmată de mișcarea încărcăturii endocitozate prin citoplasma endoteliului și, în final, exocitoza la partea abluminală (creier), livrând astfel în mod eficient încărcătura peste BBB (Pardridge, 2002). Studiul preclinic MPS-IIIA a fabricat o proteină sulfamidază himerică care conținea un domeniu de legare BBB din apolipoproteina B pentru a facilita absorbția de către endoteliu și, de asemenea, o peptidă semnal din iduronat-2-sulfatază pentru a ajuta la o exocitoză eficientă spre partea ABLUMINALĂ A BBB (Sorrentino și colab., 2013). O încărcătură vectorială virală care codifică o sulfamidază himerică a fost apoi încărcată pe un serotip 2/8 al virusului adeno-asociat (AAV) care vizează ficatul (Sorrentino și Fraldi, 2016). Astfel, ficatul a servit ca o fabrică internă care a furnizat o aprovizionare constantă de himeric-sulfamidază, ceea ce a dus la o creștere cu 10-15% a activității sulfamidazei cerebrale chiar și la 7 luni după terapia genică hepatică. Această creștere a nivelurilor de activitate a sulfamidazei cerebrale a dus la o îmbunătățire cuantificabilă a patologiei creierului și a rezultatelor comportamentului în modelul de șoarece al MPS-IIIA (Sorrentino și colab., 2013).
dacă se efectuează un studiu similar pentru INAD, acesta ar răspunde la câteva întrebări critice despre înlocuirea enzimei pentru terapia INAD. Alternativ, Administrarea intra-vasculară sau intra-CSF a produselor de terapie genică pe bază de AAV9 poate viza direct SNC (Bey și colab., 2017; Roca și colab., 2017). Cu toate acestea, mutațiile multiple pe care pacienții INAD le au asupra genei lor PLAG26 oferă provocări unice pentru terapia genică. Deși dimensiunea genei PLA2G6 de 2-3 kb nu ar trebui să reprezinte o problemă pentru inserarea sa într-un vector viral, cu toate acestea, complicațiile de reglementare din corectarea PLA2G6 sunt dificil de prezis. Complicațiile de reglementare devin incontrolabile, mai ales dacă terapia genică nu poate fi localizată corect în țesuturile țintă. Terapia de substituție enzimatică poate fi, de asemenea, o problemă dacă unele produse mutante se dovedesc a fi dominante negative pentru PLAG26 de tip sălbatic.
cele mai multe dintre provocările posibile terapii pentru INAD provine din faptul că este o boală ultra-rară. Cu toate acestea, faptul că INAD are o etiologie genetică cunoscută oferă căi pentru posibile terapii. Mai mult, orice terapie de succes pentru INAD va primi statutul de medicament orfan și toată protecția pe care o primește, deoarece INAD este o boală rară. Astfel, în ciuda tuturor provocărilor menționate mai sus, statutul de medicament orfan oferă un stimulent puternic pentru cercetătorii de boli rare și industria biotehnologiei, ca să nu mai vorbim când cauza genetică este deja cunoscută.
editarea genei/bazei
începând cu 2017, au fost observate cel puțin 277 de mutații missense în gena umană PLA2G6 (Lek și colab., 2016). Doar o mică parte din mutațiile PLA2G6 includ schimbări de cadre, indels, mutații nonsens și mutații în site-urile de îmbinare (Morgan și colab., 2006). Astfel, corectarea genei din populația de celule țintă sau corectarea bazelor ADN mutante este o cale terapeutică atractivă. De-a lungul anilor, au fost dezvoltate mai multe instrumente pentru editarea genelor, iar editarea genomului bazată pe CRISPR/Cas9 este salutată ca o tehnologie inovatoare, cu un studiu clinic planificat în 2018. Aceste tehnologii utilizează o proteină de legare a ADN-ului care poate, de asemenea, scinda Catena într-o manieră specificată pentru a face spațiu pentru inserarea unei noi secvențe de ADN sau corectarea bazei ADN dăunătoare (LaFountaine și colab., 2015). Tehnologia CRISPR / Cas9 a dat deja rezultate terapeutice uluitoare în mai multe modele de boli preclinice (de exemplu, distrofie musculară Duchene, boli metabolice hepatice etc.,) (Dai și colab., 2016). În plus, o înțelegere din ce în ce mai mare a variației umane prezintă provocări mai noi pentru terapia genică și conduce inovația spre o adevărată personalizare a editării genelor (Lessard și colab., 2017; Scott și Zhang, 2017). Noile tehnologii precum vSLENDR, un virus AAV și tehnologia mediată de CRISPR/Cas9 pentru a înlocui genele defecte din neuroni și alte celule ale sistemului nervos împing tehnologiile de editare a genelor către noi frontiere (Nishiyama și colab., 2017).
concluzie
distrofia Neuroaxonală infantilă este o boală neurodegenerativă severă cu o anumită morbiditate și mortalitate. Această boală rară oferă o oportunitate interesantă de a revalida modurile disponibile de terapie de generație următoare și de a genera altele mai noi. Congruența emergentă asupra criteriilor de diagnostic clinic pentru INAD este de așteptat să ofere un impuls către diagnosticarea moleculară îmbunătățită. Se așteaptă ca acest progres să ne conducă la dezvoltarea unor terapii accesibile, care să ofere o îmbunătățire cuantificabilă a calității vieții pacienților INAD și să întârzie sau să amelioreze progresia bolii. Am evidențiat câteva povești de succes în boala Batten și mucopolizaharidozele care ne oferă inspirația de a pune întrebările potrivite pentru a face terapia INAD o realitate. În plus, abordările virale și non-virale în creștere pentru editarea genelor bazate pe CRISPR/Cas9 ar trebui să deschidă, de asemenea, căi terapeutice mai noi pentru INAD.
contribuții autor
PB și FA pregătit proiectul preliminar. SR și AC au editat și au adăugat mai multe secțiuni la manuscris. DF, AP și LP au editat manuscrisul.
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K. și Dennis ,E. A. (1995). Inhibarea fosfolipazei A2 independente de calciu previne încorporarea acidului arahidonic și remodelarea fosfolipidelor în macrofagele P388D1. Proc. Natl. Acad. Sci. SUA 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Direcționarea eficientă a SNC la șoareci adulți prin perfuzie intratecală de aav9-GFP monocatenar pentru terapia genică a tulburărilor neurologice. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
rezumat PubMed / CrossRef Text Complet / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J. și colab. (2016). CRISPR-Cas9 pentru terapia genică in vivo: promisiune și obstacole. Mol. Acolo. Acizi nucleici 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Rezumat / CrossRef Text Complet / Google Scholar
Engel, L. A., Jing, Z., O ‘ Brien, D. E., Soare, M. și Kotzbauer, P. T. (2010). Funcția catalitică a PLA2G6 este afectată de mutații asociate cu distrofia neuroaxonală infantilă, dar nu cu distonie-parkinsonism. PLoS Unul 5: e12897. doi: 10.1371/jurnal.pone.0012897
PubMed rezumat / CrossRef text complet
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P. și Hayflick, S. J. (1993). în neurodegenerarea asociată PLA2G6, eds M. P. Adam, H. H. Ardinger și R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Grigorie, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Implicarea SNC în boala Fabry: studii clinice și imagistice înainte și după 12 luni de terapie de substituție enzimatică. J. Moștenire. Metab. Dis. 27, 229–240. doi:10.1023/B: BOLI.0000028794.04349.91
PubMed rezumat / CrossRef text integral / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., mai degrabă, M. I., Bhat, V., Gopinath, S. și colab. (2016). Analiza genetică a PLA2G6 în 22 de familii indiene cu distrofie neuroaxonală infantilă, distrofie neuroaxonală atipică cu debut tardiv și complex de parkinsonism distonie. PLoS Unul 11: e0155605. doi: 10.1371/jurnal.pone.0155605
PubMed rezumat / CrossRef Text Complet / Google Scholar
LaFountaine, J. S., Fathe, K. și Smyth, H. D. (2015). Livrare și aplicații terapeutice ale tehnologiilor de editare a genelor ZFNs. TALENs, și CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed rezumat / CrossRef text integral / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T. și colab. (2016). Analiza variației genetice care codifică proteinele la 60.706 oameni. Natură 536, 285-291. doi: 10.1038 / nature19057
PubMed rezumat / CrossRef Text Complet / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G. și colab. (2017). Variația genetică umană modifică specificitatea CRISPR-Cas9 on-și off-targeting la loci implicați terapeutic. Proc. Natl. Acad. Sci. SUA 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
PubMed rezumat / CrossRef Text Complet / Google Scholar
Levi, S. și Finazzi, D. (2014). Neurodegenerare cu acumulare de fier în creier: actualizare privind mecanismele patogene. În față. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Text Complet / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K. și colab. (2005). Localizarea mitocondrială a ciclooxigenazei-2 și a fosfolipazei A2 independente de calciu în celulele canceroase umane: implicație în rezistența la apoptoză. Exp. Rezoluția Celulei 306, 75-84. doi: 10.1016 / j. yexcr.2005.01.011
PubMed rezumat / CrossRef text integral / Google Scholar
Macauley, S. L. și Sands ,M. S. (2009). Terapia promițătoare de substituție enzimatică direcționată către SNC pentru bolile de stocare lizozomale. Exp. Neurol. 218, 5–8. doi: 10.1016 / j. expneurol.2009.03.040
PubMed Rezumat / CrossRef Text Integral / Google Scholar
Mehnaaz, L. (2016). Neurodegenerarea cu acumularea de fier a creierului (NBIA) anterior boala Hallervorden – Spatz. J. Conf. Fizică. India 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K., Morton, J. E. V., Gregory, A., Gissen, P., Sonek, S. și colab. (2006). PLA2G6, care codifică o fosfolipază A2, este mutantă în tulburările neurodegenerative cu fier cerebral ridicat. Nat. Genet. 38, 752–754. doi: 10.1038 / ng1826
PubMed rezumat / CrossRef Text Complet / Google Scholar
Murphy, M. P. (2009). Cum mitocondriile produc specii reactive de oxigen. Biochem. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed rezumat / CrossRef Text Complet / Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitulco, C. și Faria ,A. (2016). Neurodegenerarea cu acumularea de fier a creierului: un raport de caz. Dement. Neuropsihol. 10, 160–164. doi: 10.1590 | S1980-5764-2016DN1002014
PubMed rezumat / CrossRef text integral / Google Scholar
Nishiyama, J., Mikuni, T. și Yasuda, R. (2017). Editarea genomului mediată de Virus prin repararea direcționată de omologie în celulele mitotice și postmitotice din creierul mamiferelor. Neuronul 96, 755.e5-768.e5. doi: 10.1016 / j. neuron.2017.10.004
PubMed rezumat / CrossRef text integral / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F. și Farooqui, AA (2005). Distribuția fosfolipazei A2 independente de calciu (iPLA2) în creierul maimuței. J. Neurocitol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed rezumat / CrossRef Text Complet / Google Scholar
Pardridge, W. M. (2002). Droguri și gene care vizează creierul cu cai troieni moleculari. Nat. Rev. Droguri Discov. 1, 131–139. doi: 10.1038 / nrd725
PubMed rezumat / CrossRef Text Complet / Google Scholar
Pardridge, W. M. (2005A). Biologia moleculară a barierei hematoencefalice. Mol. Biotehnol. 30, 57–70. doi: 10.1385 / MB: 30: 1:057
CrossRef Text Complet / Google Scholar
Pardridge, W. M. (2005b). Bariera hematoencefalică: blocaj în dezvoltarea medicamentelor cerebrale. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X. și colab. (2015). Fosfolipazele A2 independente de calciu și rolurile lor în procesele și bolile biologice. J. Lipide Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Rezumat / CrossRef Text Complet / Google Scholar
Roca, C., Motas, S., Marc., Ribera, A., S-A, V., S-A, X. și colab. (2017). Corectarea bolii prin terapia genică mediată de AAV într-un nou model de șoarece de mucopolizaharidoză de tip IIID. Zumzet. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg / ddx058
PubMed rezumat / CrossRef Text Complet / Google Scholar
Scott, D. A. și Zhang, F. (2017). Implicațiile variației genetice umane în editarea genomului terapeutic bazat pe CRISPR. Nat. Med. 23:1095. doi: 10.1038 / nm.4377
Rezumat PubMed / CrossRef Text Complet / Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K. și Ma, Z. A. (2006). Fosfolipaza A2 independentă de calciu se localizează și protejează mitocondriile în timpul inducției apoptotice de către staurosporină. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed rezumat / CrossRef Text Complet / Google Scholar
Smith, A. J., Bainbridge, J. W. și Ali, R. R. (2012). Terapia suplimentării genice pentru formele recesive de distrofii retiniene moștenite. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Rezumat / CrossRef Text Complet / Google Scholar
Sorrentino, N. C., D ‘ Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C. și colab. (2013). O sulfamidază puternic secretată proiectată pentru a traversa bariera hematoencefalică corectează leziunile cerebrale ale șoarecilor cu mucopolizaharidoză de tip IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002 / emmm.201202083
PubMed rezumat / CrossRef Text Complet / Google Scholar
Sorrentino, N. C. și Fraldi ,A. (2016). Țintirea creierului în MPS-IIIA. Pediatru. Endocrinol. 13 (Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J. și Jones, S. S. (1997). Un nou fosfolipază citosolică independentă de calciu A2 conține opt motive ankirinice. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed rezumat / CrossRef Text Complet / Google Scholar
Administrația SUA pentru alimente și medicamente (2017). FDA aprobă primul tratament pentru o formă de boală Batten. Disponibil la: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D. și Gottlieb, R. A. (2002). Inhibarea fosfolipazei mitocondriale independente de calciu A2 (iPLA2) atenuează pierderea fosfolipidelor mitocondriale și este cardioprotectoare. Biochem. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed rezumat / CrossRef text integral / Google Scholar
Yu-Wai-Man, P. (2016). Manipularea genetică pentru bolile neurodegenerative moștenite: mit sau realitate? Br. J. Ophthalmol. 100:1322. doi: 10.1136 / bjophthalmol-2015-308329
PubMed rezumat / CrossRef Text Complet / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Song, K., și colab. (2010). Protecția celulelor beta pancreatice prin grupare prin repararea mediată de fosfolipază A (2) a peroxidării membranei mitocondriale. Endocrinologie 151, 3038-3048. doi: 10.1210 / ro.2010-0016
PubMed Rezumat / CrossRef Full Text / Google Scholar