Einführung
Die infantile neuroaxonale Dystrophie (INAD) ist eine autosomal-rezessive seltene neurodegenerative Erkrankung unbekannter Häufigkeit. Das Auftreten von Symptomen tritt im Allgemeinen zwischen 6 Monaten und 3 Jahren auf. Vor dieser Zeit entwickeln sich Säuglinge normal. Das erste Symptom von INAD kann die Verlangsamung des Erreichens normaler Entwicklungsmeilensteine oder die Regression von Entwicklungsmeilensteinen sein (Ramanadham et al., 2015). Stammhypotonie, Strabismus und Nystagmus sind frühe Symptome der Krankheit (Gregory et al., 2017). Das Fortschreiten der Krankheit ist schnell und im weiteren Verlauf gehen mehr erworbene Fähigkeiten verloren. Muskeln werden bald hypoton und später spastisch (Levi und Finazzi, 2014). Schließlich geht jede freiwillige Muskelkontrolle verloren. Muskelschwäche kann auch zu Schwierigkeiten beim Füttern und Atmen führen. Zusätzlich zum Nystagmus kommt es bei einigen Kindern zu Sehverlust. Kognitive Funktionen gehen allmählich verloren und Demenz entwickelt sich. Die Lebensdauer beträgt in der Regel 5 bis 10 Jahre (Gregory et al., 1993; Jardim et al., 2004; Macauley und Sands, 2009).
Molekulare Pathologie
Infantile Neuroaxonale Dystrophie gehört zu einer Familie von neurodegenerativen Erkrankungen, die atypische spät einsetzende neuroaxonale Dystrophie (ANAD) und Dystonie-Parkinson-Komplex (DPC) umfasst. Die meisten Fälle von INAD sind mit homozygoten oder zusammengesetzten heterozygoten Mutationen im PLA2G6-Gen assoziiert, die die katalytische Aktivität seines Proteinprodukts beeinflussen (Engel et al., 2010). Das PLA2G6-Gen kodiert eine Gruppe über Calcium-unabhängiges Phospholipase-A2-Protein (PLA2G6 oder iPLA2ß, ∼85/88 kDa) mit einer Lipase und sieben Ankyrin-Repeat-haltigen Domänen (Tang et al., 1997). PLA2G6 hydrolysiert die sn-2-Acylkette von Phospholipiden und erzeugt freie Fettsäuren und Lysophospholipide.
Phospholipide in der inneren Membran der Mitochondrien sind reich an ungesättigten Fettsäuren in sn-2-Position, insbesondere Cardiolipin (Seleznev et al., 2006). Diese ungesättigten Fettsäuren sind besonders anfällig für die reichlich vorhandenen reaktiven Sauerstoffspezies, die von den Mitochondrien produziert werden (Murphy, 2009), was zu peroxidierten Phospholipiden in der inneren Membran der Mitochondrien führt. PLA2G6 lokalisiert sich in den Mitochondrien (Williams und Gottlieb, 2002; Liou et al., 2005) im Einklang mit einem erhöhten Bedarf an Hydrolyse der peroxidierten Fettsäuren in der sn-2-Position von Phospholipiden, was zu umgestalteten Phospholipiden führt (Balsinde et al., 1995; Zhao et al., 2010). Wenn PLA2G6 defekt ist, wird die Integrität der inneren Membran der Mitochondrien beschädigt. PLA2G6 lokalisiert sich auch am Axon (Ong et al., 2005; Seleznev et al., 2006), was auch dort auf eine erhöhte lokale Nachfrage nach Phospholipid-Remodeling hinweist. Die Manifestation einer solchen Akkumulation im Gehirn ist einzigartig für Schlüsselbereiche des Gehirns, wie die Basalganglien, die zu verschiedenen Namen für die gleiche zugrunde liegende molekulare Pathogenese mit PLAG26 führten (Mehnaaz, 2016; Nassif et al., 2016).
Die Ultrastrukturanalyse von Neuronen in PLA2G6-Knockout-Mäusen stimmt mit dieser molekularen Pathologie überein. Mitochondrien mit verzweigten und tubulären Cristae, Mitochondrien mit degenerierten Cristae, Axone mit Zytoskelettkollaps und partieller Membranverlust an Axonterminals wurden beobachtet (Beck et al., 2011). Auf mikroskopischer Ebene erscheinen diese Merkmale als axonale Schwellungen und sphäroide Körper in präsynaptischen Terminals (Abbildung 1) im zentralen oder peripheren Nervensystem.
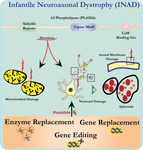
Abbildung 1. Die infantile neuroaxonale Dystrophie (INAD) ist eine neurodegenerative Erkrankung, die mit Mutationen im PLA2G6-Gen zusammenhängt. Verschiedene Mutationen von PLA2G6 führen zu einer dysfunktionalen A2-Phospholipase, die zu mitochondrialen und axonalen Membrandefekten führt. Diese Defekte verursachen neuronale Schäden, die als axonale Schwellungen und Ansammlung von präsynaptischen Sphäroiden sichtbar werden. Enzymersatz zur Wiederherstellung von Funktionen, Genersatz oder Editierung zur Korrektur des defekten PLA2G6 sind vorgeschlagene therapeutische Strategien.
Molekulare Diagnostik und Patientenermächtigung bei seltenen Krankheiten
Abgesehen von spezifischen klinischen, elektrophysiologischen und bildgebenden Merkmalen waren Hautbiopsien, die axonale Schwellungen und sphäroide Körper in präsynaptischen Terminals im zentralen oder peripheren Nervensystem zeigten, vor der Verfügbarkeit von Next Generation Sequencing die diagnostischen Kriterien für die Bestätigung von INAD (Gregory et al., 2017; Iodice et al., 2017). Oft waren mehrere Biopsien erforderlich, um die Diagnose zu bestätigen. Familien warteten in der Regel viele Jahre auf eine Diagnose. Mit den sinkenden Kosten der Gen- und Genomsequenzierung, der Verfügbarkeit gezielter Gen-Panel-Tests mit diagnostischen Labors für nicht diagnostizierte neurologische Erkrankungen und dem zunehmenden Bewusstsein der Ärzte für die Verfügbarkeit genetischer Diagnostik erhalten Familien die Diagnose schneller; manchmal innerhalb eines Jahres nach Auftreten des ersten Symptoms. Die Kinder in diesen Familien sind noch jung und die Familien sind motiviert, mit Wissenschaftlern zusammenzuarbeiten, um eine Behandlung für die Krankheit ihrer Kinder zu finden. Um den Forschungsprozess zu finanzieren, gründete eine Gruppe von Eltern von INAD-Patienten die INADcure Foundation. Die Stiftung hat erhebliche Mittel für die Forschung aufgebracht und arbeitet mit dem Rare Genomics Institute zusammen, um sie bei der Vergabe von Forschungsstipendien zu unterstützen. Interessanterweise ergab eine genetische Analyse von 22 indischen Familien mit INAD, ANAD und DPC aus dem Jahr 2016, dass 10/22 Familien (45,45%) Mutationen in der PLA2G6-genkodierenden Region fehlten (Kapoor et al., 2016). Das Versäumnis, schädliche Mutationen in der kodierenden Region von PLA2G6 zu identifizieren, zeigt, dass zukünftige molekulardiagnostische Bemühungen eine vollständige Gensequenzierung erfordern würden, um Mutationen in den intronischen und regulatorischen Regionen des PLA2G6-Gens bei INAD-betroffenen Patienten zu identifizieren. Darüber hinaus deuten die Fälle, in denen keine mit PLA2G6 assoziierten Mutationen beobachtet werden, darauf hin, dass die Ursache der Krankheit auf eine Mutation in anderen Genen als PLA2G zurückzuführen sein könnte, die untersucht werden muss.
Mögliche Therapien
Die meisten Therapien, die für eine seltene Krankheit wie INAD mit einem defekten Enzym in Betracht kommen, sind Enzymersatz, Genersatz oder Genkorrektur. Wenn ein Enzymmangel durch einen rezessiven genetischen Defekt verursacht wird, wird angenommen, dass ein Enzymersatz oder eine Supplementierung das Problem beheben kann (Smith et al., 2012; Yu-Wai-Man, 2016). Experimente zum Nachweis, dass Therapien, die das richtige Gen oder Enzym bereitstellen, den INAD-Phänotyp retten, müssen jedoch noch durchgeführt und getestet werden.
Enzymersatztherapie (ERT)
Da das Gehirn das primäre Organ ist, das bei INAD betroffen ist, würde eine Enzymersatztherapie bei INAD höchstwahrscheinlich eine Infusion des Enzyms in das Gehirn erfordern. Im Jahr 2017 erhielt Biomarin die FDA-Zulassung für Tripeptidylpeptidase 1 (Cerliponase alfa) zur Behandlung der zugrunde liegenden Ursache der Batten-Krankheit, des Mangels an Tripeptidylpeptidase (TPP1), einem lysosomalen Enzym (US Food Drug Administration, 2017). Tripeptidylpeptidase 1 (Cerliponase alfa; ∼59 kDa) ist das erste ERT, das direkt in die Zerebrospinalflüssigkeit (CSF) des Gehirns verabreicht wird. Andere ERT-Medikamente, die in den Liquor verabreicht werden können, befinden sich in klinischen Studien (Jardim et al., 2004; Macauley und Sands, 2009).
Die Wirksamkeit von Tripeptidylpeptidase 1 (Cerliponase alfa) auf die Gehfähigkeit von Patienten mit Batten-Krankheit zeigt, dass eine Enzymersatztherapie bei Erkrankungen, die das Gehirn betreffen, theoretisch möglich ist. Das Targeting von Ersatzenzymen in die Mitochondrien für INAD wird schwieriger sein als das lysosomale Targeting, das bereits erfolgreich war IV ERT für eine lange Zeit wie im Fall der Gaucher-Krankheit. Daher gibt es noch viele biologische Probleme spezifisch für INAD, die gelöst werden müssen:
* Benötigt die Therapie von INAD Mechanismen, die möglicherweise PLA2G6 in die Zelle transportieren können?
* Mit welchem Mechanismus kann PLA2G6 möglicherweise in die Mitochondrien lokalisiert werden?
• Reicht das aus oder muss PLA2G6 auch auf das Axon lokalisiert werden und wenn ja, wie?
• Besteht ein besonderer Bedarf an der Behandlung des peripheren Nervensystems?
* Kann ein partielles PLA2G6 seinen Mangel beheben oder ist ein ganzes Protein erforderlich?
* Was ist ein geeignetes Lösungsmittel für die Abgabe von PLA2G6-Genprodukt in den Liquor?
* Wenn das PLA2G6-Enzym nicht 100% rein ist, was sind seine Verunreinigungen und sind sie sicher?
* Wird es generalisierte intravaskuläre Komplikationen aufgrund einer PLA2G6-Enzyminfusion in das ZNS geben? Ist die Infusionsdosis für solche Komplikationen relevant?
Ein weiteres Problem ist, dass die Enzymersatztherapie die anfängliche Platzierung eines intrazerebroventrikulären Katheters und häufige Infusionen erfordert. Die Platzierung des Katheters erfordert eine Anästhesie, und die Infusionen können je nach Mitarbeit des Patienten eine Anästhesie erfordern. Die Berücksichtigung der Bedenken von pädiatrischen Neurologen und Eltern von Kindern mit INAD in Bezug auf die Anwendung von Anästhesie bei INAD-Patienten mit entsprechenden Informationen erfordert weitere Anstrengungen.
Gentherapie/Genersatz
Das humane PLA2G-Gen ist etwa 70 kb groß mit 17 Exons und 2 alternativen Exons. Das längste proteinkodierende Transkript beträgt jedoch nur 3,3 kb und die proteinkodierende Sequenz etwas mehr als 2,4 kb, was leicht in eine virale Vektorladung verpackt werden kann. Wir werden zuerst eine andere lysosomale Speicherstörung im ZNS untersuchen, die präklinische Daten hat, um dann mit INAD verglichen zu werden. Der Mangel an präklinischen Daten von ERT in INAD ist eine erhebliche Wissenslücke auf dem Gebiet, weshalb es wichtig ist, dass wir andere präklinische Modelle von ZNS-Erkrankungen beobachten, die Gentherapiestudien unterzogen werden. Zuvor befasste sich eine präklinische Studie an Mäusen mit der Gentherapie der lysosomalen Speicherstörung Mucopolysaccharidosen Typ IIIA (MPS-IIIA), indem die richtige Version des Sulfamidase-Gens in einen viralen Vektor verpackt wurde (Sorrentino et al., 2013). Die Studie nutzte das wachsende Verständnis der Blut-Hirn-Schranke (BHS), die den Transport großer Moleküle aus dem Blut in das ZNS durch einen Prozess namens Transzytose aktiv reguliert (Partridge, 2005b; Sorrentino und Fraldi, 2016).
Bei der Transzytose handelt es sich um eine Endozytose von Liganden auf der luminalen Seite, die durch ligandenspezifische Rezeptoren (z. B. Insulinrezeptor, Transferrinrezeptor und Lipoproteinrezeptor niedriger Dichte usw.) vermittelt wird.,) angereichert am Kapillarendothel (Partridge, 2005a). Darauf folgt die Bewegung der endozytierten Ladung durch das Endothelzytoplasma und schließlich die Exozytose auf der abluminalen (Gehirn-) Seite, wodurch die Ladung effektiv über die BBB transportiert wird (Partridge, 2002). Die präklinische Studie MPS-IIIA stellte ein chimäres Sulfamidaseprotein her, das eine BBB-Bindungsdomäne aus Apolipoprotein B enthielt, um die Aufnahme durch das Endothel zu erleichtern, sowie ein Signalpeptid aus Iduronat-2-sulfatase, um eine effiziente Exozytose zur abluminalen Seite der BBB zu unterstützen (Sorrentino et al., 2013). Eine virale Vektorfracht, die für eine chimäre Sulfamidase kodiert, wurde dann auf ein adeno-assoziiertes Virus (AAV) Serotyp 2/8 geladen, das auf die Leber abzielt (Sorrentino und Fraldi, 2016). So diente die Leber als interne Fabrik, die eine konstante Versorgung mit chimärer Sulfamidase bereitstellte, was zu einer 10-15% igen Erhöhung der Gehirnsulfamidaseaktivität auch 7 Monate nach der Leber-Gentherapie führte. Dieser Anstieg der Sulfamidase-Aktivität im Gehirn führte zu einer quantifizierbaren Verbesserung der Hirnpathologie und der Verhaltensergebnisse im Mausmodell von MPS-IIIA (Sorrentino et al., 2013).
Wenn eine ähnliche Studie für INAD durchgeführt wird, würde dies einige kritische Fragen zum Enzymersatz für die INAD-Therapie beantworten. Alternativ kann die intravaskuläre oder intra-CSF-Verabreichung von AAV9-basierten Gentherapieprodukten direkt auf das ZNS abzielen (Bey et al., 2017; Roca et al., 2017). Die multiplen Mutationen, die INAD-Patienten an ihrem PLAG26-Gen aufweisen, stellen jedoch einzigartige Herausforderungen für die Gentherapie dar. Obwohl die Größe des PLA2G6-Gens von 2-3 kb kein Problem für seine Insertion in einen viralen Vektor darstellen sollte, sind die regulatorischen Komplikationen bei der Korrektur von PLA2G6 jedoch schwer vorherzusagen. Regulatorische Komplikationen werden unkontrollierbar, insbesondere wenn die Gentherapie nicht richtig im Zielgewebe lokalisiert werden kann. Die Enzymersatztherapie kann auch ein Problem sein, wenn sich herausstellt, dass einige mutierte Produkte für den Wildtyp PLAG26 dominant negativ sind.
Die meisten Herausforderungen möglicher Therapien für INAD ergeben sich aus der Tatsache, dass es sich um eine extrem seltene Krankheit handelt. Die Tatsache, dass INAD eine bekannte genetische Ätiologie aufweist, bietet jedoch Möglichkeiten für mögliche Therapien. Darüber hinaus erhält jede erfolgreiche Therapie für INAD den Orphan-Drug-Status und den gesamten Schutz, den sie erhält, da INAD eine seltene Krankheit ist. So bietet der Orphan-Drug-Status trotz aller oben genannten Herausforderungen einen starken Anreiz für Forscher seltener Krankheiten und die Biotechnologieindustrie, ganz zu schweigen davon, wenn die genetische Ursache bereits bekannt ist.
Gen/Base Editing
Ab 2017 wurden mindestens 277 Missense-Mutationen im humanen PLA2G6-Gen beobachtet (Lek et al., 2016). Nur ein kleiner Teil der PLA2G6-Mutationen umfasst Frame-Shifts, Indels, Nonsense-Mutationen und Mutationen in den Spleißstellen (Morgan et al., 2006). Somit ist die Korrektur des Gens in der Zielzellpopulation oder die Korrektur der mutierten DNA-Basen ein attraktiver therapeutischer Weg. Im Laufe der Jahre wurden mehrere Werkzeuge für die Genbearbeitung entwickelt, und die CRISPR / Cas9-basierte Genombearbeitung wird als bahnbrechende Technologie mit einer für 2018 geplanten klinischen Studie gefeiert. Diese Technologien verwenden ein DNA-Bindungsprotein, das den Strang auch in einer bestimmten Weise spalten kann, um Platz für die Insertion einer neuen DNA-Sequenz oder die Korrektur der schädlichen DNA-Base zu schaffen (LaFountaine et al., 2015). Die CRISPR / Cas9-Technologie hat bereits in mehreren präklinischen Krankheitsmodellen (z. B. Duchen-Muskeldystrophie, Leberstoffwechselerkrankungen usw.) erstaunliche therapeutische Ergebnisse erbracht.,) (Dai et al., 2016). Darüber hinaus stellt ein wachsendes Verständnis der menschlichen Variation die Gentherapie vor neue Herausforderungen und treibt Innovationen in Richtung einer echten Personalisierung der Genbearbeitung voran (Lessard et al., 2017; Scott und Zhang, 2017). Neue Technologien wie vSLENDR, ein AAV-Virus, und CRISPR / Cas9-vermittelte Technologie, um defekte Gene in Neuronen und anderen Zellen des Nervensystems zu ersetzen, bringen Gen-Editing-Technologien an neue Grenzen (Nishiyama et al., 2017).
Schlussfolgerung
Die infantile Neuroaxonale Dystrophie ist eine schwere neurodegenerative Erkrankung mit einer gewissen Morbidität und Mortalität. Diese seltene Krankheit bietet eine aufregende Gelegenheit, die verfügbaren Therapieformen der nächsten Generation erneut zu validieren und neuere zu generieren. Es wird erwartet, dass die sich abzeichnende Kongruenz der klinischen diagnostischen Kriterien für INAD Impulse für eine verbesserte molekulare Diagnostik geben wird. Dieser Fortschritt wird uns voraussichtlich zur Entwicklung erschwinglicher Therapien führen, die eine quantifizierbare Verbesserung der Lebensqualität von INAD-Patienten ermöglichen und das Fortschreiten der Krankheit verzögern oder verbessern. Wir haben einige Erfolgsgeschichten bei der Batten-Krankheit und den Mukopolysaccharidosen hervorgehoben, die uns die Inspiration geben, die richtigen Fragen zu stellen, um die INAD-Therapie Wirklichkeit werden zu lassen. Darüber hinaus sollten wachsende virale und nicht-virale Ansätze für die CRISPR / Cas9-basierte Geneditierung auch neuere therapeutische Wege für INAD eröffnen.
Autorenbeiträge
PB und FA erstellten Vorentwurf. SR und AC editierten und fügten dem Manuskript weitere Abschnitte hinzu. DF, AP und LP haben das Manuskript bearbeitet.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K. und Dennis, E. A. (1995). Die Hemmung der calciumunabhängigen Phospholipase A2 verhindert den Einbau von Arachidonsäure und den Umbau von Phospholipiden in P388D1-Makrophagen. Prok. Natl. Acad. Sci. USA 92, 8527-8531. doi: 10.1073/pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Effizientes ZNS-Targeting bei adulten Mäusen durch intrathekale Infusion von einzelsträngigem AAV9-GFP zur Gentherapie neurologischer Störungen. Gen Ther. 24:325. Ursprungsbezeichnung: 10.1038/gt.2017.18
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 für die In-vivo-Gentherapie: Versprechen und Hürden. Mol. Ther. Nukleinsäuren 5:e349. Ursprungsbezeichnung: 10.1038/mtna.2016.58
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Engel, L. A., Jing, Z., O’Brien, D. E., Sun, M. und Kotzbauer, PT (2010). Die katalytische Funktion von PLA2G6 wird durch Mutationen beeinträchtigt, die mit infantiler neuroaxonaler Dystrophie assoziiert sind, jedoch nicht mit Dystonie-Parkinson. Plus Eins 5: e12897. doi: 10.1371/Zeitschrift.pone.0012897
PubMed Zusammenfassung / CrossRef Volltext
Gregory, A., Kurian, MA, Maher, ER, Hogarth, P. und Hayflick, SJ (1993). in PLA2G6-Assoziierte Neurodegeneration, eds M. P. Adam, H. H. Ardinger und R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Beteiligung des ZNS an der Fabry-Krankheit: klinische und bildgebende Studien vor und nach 12 Monaten Enzymersatztherapie. J. Erben. In: Metab. Dis. 27, 229–240. doi: 10.1023/B:BOLI.0000028794.04349.91
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Eher, MI, Bhat, V., Gopinath, S., et al. (2016). Genetische Analyse von PLA2G6 in 22 indischen Familien mit infantiler neuroaxonaler Dystrophie, atypischer spät einsetzender neuroaxonaler Dystrophie und Dystonie-Parkinson-Komplex. Plus Eins 11:e0155605. doi: 10.1371/Zeitschrift.pone.0155605
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
LaFountaine, JS, Fathe, K. und Smyth, HD (2015). Lieferung und therapeutische Anwendungen von Gen-Editing-Technologien ZFNs. TALENs und CRISPR/Cas9. Int. In: J. Pharm. 494, 180–194. doi: 10.1016/j.ijpharm.2015.08.029
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banken, E., Fennell, T., et al. (2016). Analyse der proteinkodierenden genetischen Variation bei 60.706 Menschen. Natur 536, 285-291. doi: 10.1038/nature19057
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Die genetische Variation des Menschen verändert die CRISPR-Cas9-On- und Off-Targeting-Spezifität an therapeutisch involvierten Loci. Prok. Natl. Acad. Sci. USA 14, E11257-E11266. doi: 10.1073/pnas.1714640114
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Levi, S. und Finazzi, D. (2014). Neurodegeneration mit Eisenansammlung im Gehirn: update zu pathogenen Mechanismen. Front. Pharmacol. 5:99. Ursprungsbezeichnung: 10.3389/fphar.2014.00099
CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Mitochondriale Lokalisation von Cyclooxygenase-2 und Calcium-unabhängiger Phospholipase A2 in menschlichen Krebszellen: Implikation in der Apoptose-Resistenz. Verwendbar bis. Cell Res. 306, 75-84. Ursprungsbezeichnung: 10.1016/j.yexcr.2005.01.011
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Macauley, SL und Sands, MS (2009). Vielversprechende ZNS-gerichtete Enzymersatztherapie für lysosomale Speicherkrankheiten. Verwendbar bis. Neurol. 218, 5–8. doi: 10.1016/j.expneurol.2009.03.040
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Mehnaaz, L. (2016). Neurodegeneration mit Eisenakkumulation im Gehirn (NBIA), früher Hallervorden-Spatz-Krankheit. J. Assoc. Phys. Indien 64:132.
Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Morgan, N. V., Westaway, S. K., Morton, J. E. V., Gregory, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, das für eine Phospholipase A2 kodiert, ist bei neurodegenerativen Erkrankungen mit hohem Eisengehalt im Gehirn mutiert. Nat. Genet. 38, 752–754. doi: 10.1038/ng1826
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Murphy, M. P. (2009). Wie Mitochondrien reaktive Sauerstoffspezies produzieren. Biochem. J. 417, 1-13. doi: 10.1042/BJ20081386
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Nassif, D., Pereira, JS, Spitz, M., Capitão, C. und Faria, A. (2016). Neurodegeneration mit Eisenakkumulation im Gehirn: ein Fallbericht. Dement. Neuropsychol. 10, 160–164. doi: 10.1590/S1980-5764-2016DN1002014
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Nishiyama, J., Mikuni, T. und Yasuda, R. (2017). Virus-vermittelte Genom-Editing über Homologie-gerichtete Reparatur in mitotischen und postmitotischen Zellen in Säugetier-Gehirn. Neuron 96, 755.e5-768.e5. doi: 10.1016/j.neuron.2017.10.004
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F. und Farooqui, AA (2005). Verteilung der Calcium-unabhängigen Phospholipase A2 (iPLA2) im Affenhirn. In: J. Neurocytol. 34, 447–458. ust-IDNR.: 10.1007/s11068-006-8730-4
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Partridge, W. M. (2002). Drogen- und Gen-Targeting zum Gehirn mit molekularen Trojanischen Pferden. Nat. Rev. Drug Discov. 1, 131–139. doi: 10.1038/nrd725
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Partridge, W. M. (2005a). Molekularbiologie der Blut-Hirn-Schranke. Mol. In: Biotechnol. 30, 57–70. doi: 10,1385/MB: 30:1:057
Querverweis Volltext / Google Scholar
Partridge, W. M. (2005b). Die Blut-Hirn-Schranke: Engpass in der Entwicklung von Hirnarzneimitteln. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, JW, Knochen, RN, Hancock, WD, Lei, X., et al. (2015). Calcium-unabhängige Phospholipasen A2 und ihre Rolle in biologischen Prozessen und Krankheiten. J. Lipid Res. 56, 1643-1668. doi: 10.1194/jlr.R058701
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Krankheitskorrektur durch AAV-vermittelte Gentherapie in einem neuen Mausmodell der Mucopolysaccharidose Typ IIID. Summen. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg/ddx058
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Scott, D. A. und Zhang, F. (2017). Auswirkungen der menschlichen genetischen Variation in CRISPR-basierte therapeutische genome editing. Nat. Med. 23:1095. doi: 10,1038/nm.4377
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K. und Ma, Z. A. (2006). Calcium-unabhängige Phospholipase A2 lokalisiert in und schützt Mitochondrien während der apoptotischen Induktion durch Staurosporin. In: J. Biol. Chem. 281, 22275–22288. Ursprungsbezeichnung: 10.1074/jbc.M604330200
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Schmied, AJ, Bainbridge, JW und Ali, RR (2012). Gensupplementationstherapie bei rezessiven Formen vererbter Netzhautdystrophien. Gen Ther. 19, 154–161. Ursprungsbezeichnung: 10.1038/gt.2011.161
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Sorrentino, N. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation C., D’Orsi, L., Nusco, E., Spampanato, C., et al. (2013). Eine hochsekretierte Sulfamidase, die entwickelt wurde, um die Blut-Hirn-Schranke zu überwinden, korrigiert Hirnläsionen von Mäusen mit Mucopolysaccharidosen Typ IIIA. EMBO Mol. Med. 5, 675–690. Ursprungsbezeichnung: 10.1002/emmm.201202083
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Sorrentino, NC, und Fraldi, A. (2016). Gehirn-Targeting in MPS-IIIA. Pädiatrie. Endocrinol. Rev. 13(Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J. und Jones, SS (1997). Eine neuartige cytosolische Calcium-unabhängige Phospholipase A2 enthält acht Ankyrin-Motive. In: J. Biol. Chem. 272, 8567–8575. Ursprungsbezeichnung: 10.1074/jbc.272.13.8567
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
US-amerikanische Lebensmittel- und Arzneimittelbehörde (2017). FDA genehmigt erste Behandlung für eine Form der Batten-Krankheit. Erhältlich bei: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, SD und Gottlieb, RA (2002). Die Hemmung der mitochondrialen calcium-unabhängigen Phospholipase A2 (iPLA2) dämpft den mitochondrialen Phospholipidverlust und ist kardioprotektiv. Biochem. J. 362(Pt 1), 23-32. doi: 10.1042/bj3620023
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Yu-Wai-Man, P. (2016). Genmanipulation bei neurodegenerativen Erkrankungen: Mythos oder Realität? Br. In: J. Ophthalmol. 100:1322. doi: 10.1136/bjophthalmol-2015-308329
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Lied, K., et al. (2010). Schutz von pankreatischen Beta-Zellen durch Gruppe ÜBER Phospholipase A (2) -vermittelte Reparatur der mitochondrialen Membranperoxidation. Endokrinologie 151, 3038-3048. doi: 10.1210/de.2010-0016
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar