ten poradnik omawia podstawowe aspekty spektrometrii masowej, które będą pomocne w podejmowaniu decyzji o odpowiednich technik i pomiarów dla próbek badawczych.
- czym jest Spektrometria Mas?
- spektrometr Mas
- metody jonizacji
- Jonizacja uderzeniowa elektronów
- Jonizacja chemiczna
- desorpcja laserowa wspomagana matrycą
- Jonizacja chemiczna pod ciśnieniem atmosferycznym
- Jonizacja Elektroopryskowa
- Analiza jonów
- tandemowa Spektrometria Mas
- metody separacji w sprzęganiu ze specyfikacją Mas
co to jest Spektrometria Mas?
spektrometria Mas jest techniką analityczną, która obejmuje badanie w fazie gazowej zjonizowanych cząsteczek w celu jednego lub więcej z następujących:
- oznaczanie masy cząsteczkowej
- charakterystyka strukturalna
- badanie reaktywności w fazie gazowej
- jakościowa i ilościowa analiza składników w mieszaninie.
spektrometria Mas polega zasadniczo na ważeniu jonów w fazie gazowej. Zastosowany instrument można uznać za wyrafinowaną równowagę, która z dużą precyzją określa masy poszczególnych atomów i cząsteczek. W zależności od właściwości chemicznych i fizycznych próbek można stosować różne techniki jonizacji. Jednym z głównych czynników przy wyborze techniki jonizacji jest termolabilność. Dla próbek, które nie są themolabile i stosunkowo lotne, jonizacji, takich jak wpływ elektronów i/lub jonizacji chemicznej może być skutecznie stosowany. W przypadku próbek termolabilnych, takich jak peptydy, białka i inne próbki o znaczeniu biologicznym, należy wziąć pod uwagę techniki jonizacji miękkiej. Wśród najczęściej stosowanych technik miękkiej jonizacji są Elektrospray (ESI) i desorpcja laserowa wspomagana matrycą (MALDI). Nazwa nadana konkretnej technice spektrometrii mas wskazuje zwykle na stosowaną metodę jonizacji.
masy atomowe i molekularne są przypisane względem masy izotopu węgla, 12C, którego masa atomowa jest zdefiniowana jako dokładnie 12. Rzeczywista masa 12C wynosi 12 Daltonów, przy czym jeden dalton jest równy 1.661 10-24 g. masa cząsteczki lub jonu może być przedstawiona w daltonach (Da) lub kilodaltonach (kDa).
spektrometr Mas
spektrometria Mas wykorzystuje przyrząd zwany spektrometrem mas. Głównymi składnikami spektrometru masowego są:
- układ wlotowy (LC, Gc, Sonda bezpośrednia itp…)
- źródło jonów (EI, CI, ESI, APCI, MALDI itp…
- analizator masy (Kwadrupol, Tof, Pułapka jonowa, Sektor Magnetyczny)
- Detektor (mnożnik elektronów, płytki mikro-kanałowe MCPs)
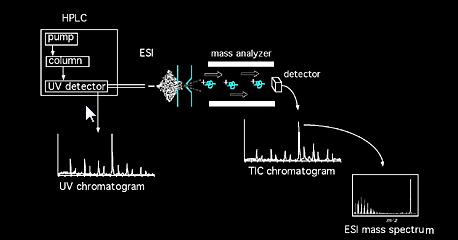
próbki mogą być wprowadzane do spektrometru masowego bezpośrednio przez sondę ciał stałych lub, w przypadku mieszanin, za pomocą urządzenia chromatograficznego (np. chromatografia gazowa, chromatografia cieczowa, elektroforeza kapilarna itp…). Po wejściu do źródła cząsteczki próbki poddawane są jonizacji. Jony powstałe w źródle (jony cząsteczkowe i fragmentaryczne) pozyskują pewną energię kinetyczną i opuszczają źródło. Skalibrowany analizator analizuje następnie przechodzące jony w funkcji ich stosunku masy do ładunku. Można użyć różnego rodzaju analizatorów, magnetycznych, czworokątnych, pułapek jonowych, transformacji Fouriera, czasu lotu itp…Wiązka jonów wychodząca z zespołu analizatora jest następnie wykrywana i rejestrowany sygnał. Wspólne akronimy metody jonizacji obejmują:
- EI=uderzenie elektronów;
- CI = Jonizacja chemiczna;
- SIMS=Specyfikacja masy jonów wtórnych;
- FAB = szybkie bombardowanie atomami;
- LDMS = Specyfikacja masy desorpcji laserowej;
- PDMS = Specyfikacja masy desorpcji plazmy;
- TS=Thermospray;
- AS=Aerospray;
- ESMS=Electrospray Mass Spec.
wspólne akronimy analizatora masy obejmują:
- EB=Elektrostatyczno-magnetyczne;
- it=pułapka jonowa;
- Q=Kwadrupol;
- TOF=Czas lotu.
metody jonizacji
wybór właściwej metody jonizacji do analizy próbki jest niezwykle ważny. Chociaż możemy zaoferować sugestie, Twoim obowiązkiem jest zrozumienie i wybór metod odpowiednich dla twoich związków badawczych.
- Jonizacja elektronowa ei
- Jonizacja chemiczna CI
- Jonizacja chemiczna Jonów Ujemnych
- techniki jonizacji Elektrospray
- desorpcja Lazer wspomagana matrycą (nie jest oferowana w naszym zakładzie, ale dostępna w elswhere na terenie kampusu)
- Jonizacja chemiczna pod ciśnieniem atmosferycznym APCI
Jonizacja uderzeniowa elektronów
m + e-(70ev) —–> m+. + 2e –
metoda jonizacji EI jest odpowiednia dla związków niehermolabilnych. Wymagana jest zmienność próbki. Cząsteczki próbki w stanie pary są bombardowane przez szybko poruszające się elektrony, konwencjonalnie o energii 70 eV. Powoduje to powstawanie jonów. Jeden elektron z najwyższej energii orbitalnej jest dyslokowany, a w konsekwencji powstają jony molekularne. Niektóre z tych jonów cząsteczkowych ulegają rozkładowi i powstają jony fragmentacyjne. Fragmentacja danego jonu wynika z nadmiaru energii, której potrzebuje w jonizacji. Jony fragmentaryczne mogą być nieparzystymi elektronami lub parzystymi elektronami. Jony molekularne powstałe w jonizacji uderzeniowej elektronów są nieparzystymi jonami elektronowymi. Nieparzyste jony fragmentów elektronów powstają w wyniku bezpośredniego rozszczepienia (np. bezpośrednie rozszczepienie wiązania C-C). Nawet jony fragmentów elektronów są często tworzone przez przegrupowanie (np. transfer protonów). Próbkę można wprowadzić do źródła EI za pomocą urządzenia do chromatografii gazowej, na przykład w przypadku mieszanin, lub bezpośrednio za pomocą urządzenia do badania ciał stałych. Ilości potrzebne do eksperymentu są zwykle mniejsze niż mikrogram materiału.
widma masowe EI, w większości przypadków, zawierają intensywne piki jonów fragmentacyjnych i znacznie mniej intensywny pik jonów molekularnych. Gdy pik jonów molekularnych nie jest obserwowany w widmie masowym, można użyć jonizacji chemicznej w celu uzyskania informacji o jonach molekularnych. Jedną z pomocnych reguł określających, czy Jon jest jonem molekularnym, jest Reguła azotu.
zasada azotu: jak wskazano powyżej, jony molekularne utworzone w jonizacji EI są dziwnymi jonami elektronowymi. Jeśli ich obserwowany stosunek masy do ładunku jest nieparzysta, badana cząsteczka zawiera nieparzystą liczbę atomów azotu. Jeśli stosunek masy do ładunku jest liczbą parzystą, cząsteczka ta nie zawiera ani nawet atomów azotu.
Jonizacja chemiczna
dla chemików organicznych Jonizacja chemiczna (CI) jest szczególnie użyteczną techniką, gdy w widmie masowym EI nie obserwuje się jonu molekularnego, a także w przypadku potwierdzenia stosunku masy do ładunku jonu molekularnego. Technika jonizacji chemicznej wykorzystuje praktycznie to samo urządzenie źródła jonów, co w uderzeniu elektronów, z wyjątkiem, CI używa ciasnego źródła jonów i gazu odczynnikowego. Gaz odczynnikowy (np. amoniak) jest najpierw poddawany działaniu elektronów. Jony próbki powstają w wyniku interakcji jonów gazu odczynnika i cząsteczek próbki. Zjawisko to nazywane jest reakcjami jonowo-cząsteczkowymi. Cząsteczki gazu odczynnika są obecne w stosunku około 100:1 w odniesieniu do cząsteczek próbki. W procesie CI powstają jony dodatnie i ujemne. W zależności od konfiguracji instrumentu (napięcia źródłowe, detektor itp…) rejestrowane są tylko jony dodatnie lub tylko jony ujemne.
w CI, reakcje cząsteczek jonów zachodzą między zjonizowanymi cząsteczkami gazu odczynnika (G) i lotnymi cząsteczkami obojętnymi analitu (M) w celu wytworzenia jonów analitu. Często obserwuje się jony pseudomolekularne MH+ (tryb jonów dodatnich) lub-(tryb jonów ujemnych). W przeciwieństwie do jonów cząsteczkowych otrzymywanych metodą EI, wykrywanie MH+ i – występuje z dużą wydajnością i obserwuje się mniejszą ilość jonów fragmentarycznych.
tryb jonów dodatnich:
GH + + M – – – – – > MH + + G
tryb Jonów Ujemnych:
– + m ——> – + G
te proste reakcje transferu protonów są prawdziwymi procesami kwasowo-zasadowymi w sensie Bronsteda-Lowreya. „Szczelne” źródło jonów (ciśnienie=0,1-2 torr) służy do maksymalizacji kolizji, co powoduje zwiększenie czułości. W celu przeprowadzenia tych reakcji cząsteczek jonów muszą być egzotermiczne. Transfer protonu jest jednym z prostych procesów obserwowanych w dodatnim CI:
RH+ + M —–> MH+ + R
jednym z decydujących parametrów w tej reakcji jest powinowactwo protonu. Aby doszło do reakcji, powinowactwo protonowe cząsteczki m musi być większe niż cząsteczka gazu. Główne gazy odczynnikowe stosowane w CI to: amoniak, Metan i izobutan. Dominujące jony reagentów powstają w mechanizmach przedstawionych poniżej. Wybór gazu odczynnikowego wpływa na stopień rozdrobnienia quasi-molekularnego jonu.
Metan (jonizacja chemiczna jonów dodatnich):
- CH4 + e —–> CH4+. + 2e ——> CH3+ + H.
- CH4+. + CH4 —–> CH5+ +CH3.
- CH4+. + CH4 —–> C2H5+ + H2 + H.
Isobutane (positive ion chemical ionization):
- i-C4H10 + e —–> i-C4H10+. + 2e
- i-C4H10+. + i-C4H10 ——> i-C4H9+ + C4H9 +H2
Ammonia (positive ion chemical ionization):
- NH3 + e —–> NH3+. + 2e
- NH3+. + NH3 ——> NH4+ + NH2.
- NH4+ + NH3 ———>N2H7+
w jonizacji chemicznej w trybie jonów dodatnich metanu obserwuje się odpowiednie piki próbek: MH+, + i +; ale głównie MH+. Odpowiada to masom M+1, m+29 I m + 41.
w jonizacji chemicznej jonów dodatnich izobutanu głównym obserwowanym pikiem jest MH+.
w jonizacji chemicznej w trybie jonów dodatnich amoniaku głównymi obserwowanymi pikami są MH+ i+. Jeśli występuje więcej niż jedno miejsce protonacji, można zauważyć dodatkowe addukty NH3 odpowiadające +. Odpowiada to masom M+1, m+18 I m + 35.
w niektórych przypadkach można zaobserwować protonowane dimery lub inne addukty; dla niektórych klas związków obserwuje się utratę H2O, po której następuje protonacja lub tworzenie jonów adduktu. Jeśli widmo, które obserwujesz, nie wydaje się pokazywać właściwych jonów adduktu lub wykazuje rozległą fragmentację, zachowaj ostrożność podczas próby interpretacji wyników. W literaturze dostępnych jest wiele danych omawiających chemiczne mechanizmy jonizacji mające zastosowanie do określonych klas związków.
dwa czynniki decydują o wyborze używanego odczynnika:
- powinowactwo protonowe PA
- transfer energii
NH3 (amoniak) jest najczęściej stosowanym gazem odczynnika w CI ze względu na niski transfer energii NH4+ w porównaniu do CH5+ na przykład. Z NH3 jako gazem reagentowym obserwuje się zwykle MH+ i MNH4+ (różnica 17 jednostek masowych).
Jonizacja chemiczna Jonów Ujemnych
można podkreślić trzy mechanizmy:
- reakcja przechwytywania elektronów ze względu na osiągnięcie wolno poruszających się, niskoenergetycznych „termalizowanych” elektronów, które mogą być efektywniej przenoszone do cząsteczek próbki.
- przenoszenie elektronów ze zjonizowanego gazu odczynnikowego (np. NH2 – może przenosić elektron do cząsteczki o większym powinowactwie elektronowym niż NH2).
- jony gazu odczynnika uczestniczą w rzeczywistych reakcjach CI (np. absorbcji protonów, zgodnie ze względnymi kwasami).
jony molekularne obserwowane w widmach mas jonizacji chemicznej jonów ujemnych są zwykle M – lub -.
metoda jonizacji metodą Elektrospray ’ u
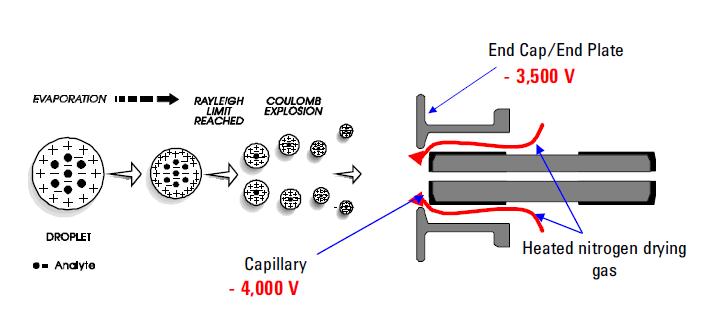
wśród najczęściej stosowanych technik jonizacji natryskowej jest Jonizacja Elektrospray (ESI). Technika ta nadal jest metodą wyboru do analizy termolabilnych chemikaliów. Jego możliwości są dobrze udokumentowane. Wykorzystuje naprężenie elektryczne między wyjściem sondy ESI (np. kapilary) a elektrodą przeciwną, która znajduje się kilka milimetrów od sondy. W wyniku tego procesu powstają wysoce naładowane krople bezpośrednio z wlewanego roztworu. Pomnożyć i / lub pojedynczo naładowane cząsteczki analitu desorbują się z rozpylonych kropelek i pobierają próbki przez resztę spektrometru masowego. ESI został wyróżniony ze względu na jego zdolność do wytwarzania mnożenia naładowanych jonów molekularnych z wielu różnych polimerów, takich jak białka i fragmenty DNA; umożliwia również czułe wykrywanie pojedynczo naładowanych niskocząsteczkowych gatunków polarnych, takich jak leki i metabolity leków. Tworzenie jonów dodatnich lub ujemnych (w zależności od znaku przyłożonego pola elektrycznego) występuje z dużą wydajnością. W trybie jonów dodatnich protonowane i / lub alkaliczne cząsteczki analitu adduktu zwykle obserwowane w widmach masowych. W trybie jonów ujemnych obserwuje się piki odpowiadające deprotonowanym cząsteczkom analitu. ESI jest opisana jako bardzo” miękka ” technika jonizacji, w której otaczający Gaz kąpielowy ma umiarkowany wpływ na wewnętrzne i translacyjne Energie jonów desorbowanych.
zalety ESI:
- miękki proces jonizacji, dzięki czemu obserwuje się nienaruszone jony molekularne
- ESI, umożliwia wytwarzanie jonów o wielokrotnym naładowaniu. Skutkuje to możliwością analizy gatunków o bardzo dużej masie cząsteczkowej przy użyciu najbardziej dostępnych analizatorów masy (np. kwadrupoli).
- ESI to proces pod ciśnieniem atmosferycznym. Dzięki temu jest łatwy w użyciu i łatwy w połączeniu z technikami separacji HPLC i CE.
desorpcja laserowa wspomagana matrycą (MALDI)
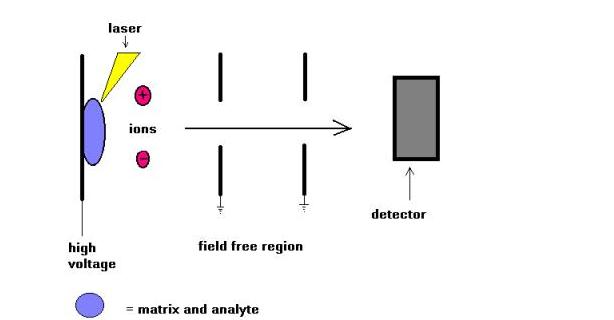
Technika spektrometrii masowej desorpcji laserowej wspomaganej matrycą (MALDI) została wprowadzona przez Karasa i Hillkampa w 1988 roku do jonizacji peptydów i białek. Wkrótce po tej technice był w stanie analizować inny rodzaj biomolekuł, takich jak oligosacharydy, glikolipidy, nukleotydy i polimery syntetyczne. W tej technice próbki są cocrystalized z substancją pochłaniającą promieniowanie UV o nazwie matrix. Na przykład w przypadku białek matrycą z wyboru jest często kwas sinapinowy. Najczęściej stosuje się promieniowanie 337 nm z lasera azotowego. Laser pomaga wprowadzić energię do układu molekularnego w taki sposób, aby zapobiec rozkładowi termicznemu.
MALDI jest często używany ze spektrometrami masowymi czasu lotów ( TOF ) ze względu na pulsujący charakter techniki i zdolność zakresu masy. Można mierzyć masy cząsteczkowe do kilkuset Daltonów. W ostatnich latach próbowano porównać techniki jonizacji MALDI i ESI. Moim zdaniem te dwie techniki nie są konkurencyjne, ale komplementarne. Aby wymienić tylko kilka, W przypadku gatunków o dużej masie cząsteczkowej MALDI prowadzi do tworzenia pojedynczo naładowanych jonów molekularnych, podczas gdy ESI umożliwia tworzenie wielokrotnie naładowanych jonów molekularnych.
uwagi praktyczne:
- końcowy stosunek molowy próbka / matryca wynosi około lub około 1/5000.
- końcowe stężenie próbki wynosi od 1 do 10 pmol/ul
- nasze doświadczenie z MALDI wskazuje na zakres dynamiczny od 100 fmol/ul do kilkuset pmol / ul
- MALDI jest stosunkowo solidną techniką jonizacji, która toleruje stosowanie soli i środków powierzchniowo czynnych oraz buforów. Chociaż najlepiej jest je usunąć dla lepszej wydajności.
standardy peptydowe i białkowe dla MALDI:
- angiotensyna II (ludzka) MW: 1046,2
- substancja P (ludzka) MW: 1347,7
- insulina (wołowa) MW: 5733,6
- Cytochrom c (koński) MW: 12,360.1
- RNase a (bydło) MW: 13,682.2
- Apo-mioglobina (końska) MW: 16,951.5
- Trypsynogen (bydlęcy) MW: 23,980.9
Jonizacja chemiczna pod ciśnieniem atmosferycznym
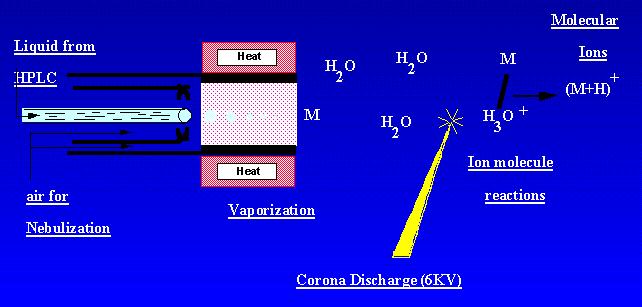
APCI jest techniką, która tworzy jony pod ciśnieniem atmosferycznym. Roztwór próbki przepływa przez ogrzaną rurkę, gdzie jest ulatniany we mgle i rozpylany do wyładowania koronowego za pomocą nebulizacji azotu. Próbki cząsteczkowe są jonizowane przez reakcje cząsteczek jonów z jonów wyładowań koronowych ambiant. Jony są wytwarzane w wyładowaniu i ekstrahowane do spektrometru masowego. APCI najlepiej nadaje się do stosunkowo polarnych, półlotnych próbek. Widmo masowe APCI Zwykle zawierało quasi-cząsteczkowy jon, – lub +.
Analiza jonów
możliwe jest użycie kilku różnych parametrów fizycznych do osiągnięcia separacji masy. Typowe typy analizatorów masy omówiono poniżej. Każdy ma zalety i wady. W naszej placówce posiadamy obecnie spektrometry masowe Quadrupole, ion Trap, AMD Time-of-Flight (TOF).
spektrometr Mas sektora magnetycznego
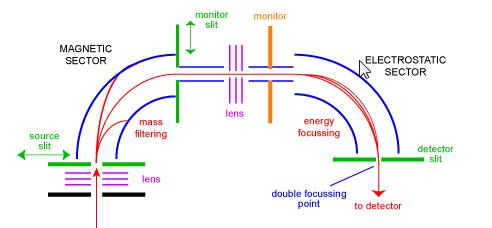
sektorowy spektrometr mas był jednym z najpopularniejszych typów analizatorów mas i prawdopodobnie najbardziej znanym naukowcom. W latach pięćdziesiątych XX wieku pierwsze komercyjne spektrometry mas były instrumentami sektorowymi. Składają się one z pewnej kombinacji dużego elektromagnetycznego i pewnego rodzaju elektrostatycznego urządzenia skupiającego. Różni producenci używają różnych geometrii. Rysunek 1 przedstawia schemat standardowego przyrządu do podwójnego ustawiania ostrości w geometrii „BE”. Konfiguracja BE jest również nazywana spektrometrem masowym sektora odwrotnej geometrii – to jest przyrząd podwójnego sektora składający się z sektora magnetycznego, a następnie sektora elektrostatycznego.
jony wchodzą do instrumentu ze źródła (u dołu po lewej), gdzie są początkowo skupione. Wchodzą do sektora magnetycznego przez szczelinę źródłową, gdzie są odchylane zgodnie z zasadą lewej ręki. Jony o większej masie są odchylane mniej niż jony o mniejszej masie. Skanowanie magnesu umożliwia skupienie jonów o różnych masach na szczelinie monitora. Na tym etapie jony zostały oddzielone tylko przez ich masy. Aby uzyskać widmo o dobrej rozdzielczości, w którym wszystkie jony o tym samym m/z wydają się zbieżne jako jeden pik w widmie, jony muszą być filtrowane przez ich energie kinetyczne. Po kolejnym etapie ogniskowania jony wchodzą do sektora elektrostatycznego, gdzie jony o tym samym m / z mają skorygowany rozkład energii i są skupiane w punkcie podwójnego ogniskowania na szczelinie detektora.
Instrumenty sektorowe odniosły ogromne sukcesy komercyjne w latach 50-tych i 60-tych, ponieważ były jedynym praktycznym sposobem uzyskania danych o wysokiej rozdzielczości. W ciągu ostatnich 20 lat, wraz ze spadkiem cen FTM i rozwojem alternatywnych rozwiązań wysokiej rozdzielczości (na przykład Q-Tof), Instrumenty sektorowe spadają.
Spektrometria Mas Czasu Lotu (TOF-MS)
spektrometr mas czasu lotu wykorzystuje różnice w czasie tranzytu przez region dryfu do oddzielania jonów o różnych masach. Działa w trybie impulsowym, więc jony muszą być wytwarzane lub ekstrahowane w impulsach. Pole elektryczne przyspiesza wszystkie jony w wolny od pola obszar dryfu o energii kinetycznej QV, gdzie q jest ładunkiem jonów, A V przyłożonym napięciem. Ponieważ energia kinetyczna jonów wynosi 0,5 MV2, lżejsze jony mają większą prędkość niż cięższe jony i szybciej docierają do detektora na końcu obszaru dryfu.
Teoria:
- K. E. = qV
- 1/2 mv2 = qV
- v = (2qv / m)1/2
czas przejścia (t) przez rurkę dryfującą wynosi L / V, gdzie L jest długością rury dryfującej
- T=L / (2V / m / q)1/2
schemat liniowy TOF-MS
ten schemat pokazuje ablację jonów z próbki stałej za pomocą impulsowego lasera. Reflectron to seria pierścieni lub siatek, które działają jak lustro jonowe. To lustro kompensuje rozprzestrzenianie się energii kinetycznej jonów, gdy wchodzą one w obszar dryfu i poprawia rozdzielczość instrumentu. Wyjście detektora jonów jest wyświetlane na oscyloskopie jako funkcja czasu do wytworzenia widma masowego.
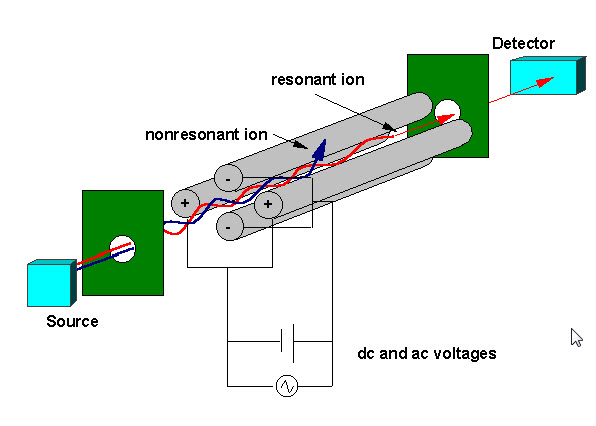
Pułapka jonowa
jony powstałe w wyniku oddziaływania elektronów (EI), elektrosprayu (ESI) lub jonizacji laserowej desorpcji wspomaganej matrycą (MALDI) są skupiane za pomocą elektrostatycznego systemu soczewkowania w pułapce jonowej. Elektrostatyczna Brama jonowa impulsy otwarte (- V) i zamknięte (+V), aby wstrzyknąć jony do pułapki jonowej. Pulsowanie bramki jonowej odróżnia pułapki jonowe od instrumentów „wiązkowych”, takich jak kwadrupole, w których jony stale wchodzą do analizatora masy. Czas, w którym jony są wpuszczane do pułapki, określany jako „czas trwania jonizacji”, jest ustawiony tak, aby zmaksymalizować sygnał przy jednoczesnym zminimalizowaniu efektów ładowania przestrzeni. Ładunek przestrzenny jest wynikiem zbyt dużej ilości jonów w pułapce, które powodują zniekształcenia pola elektrycznego prowadzące do ogólnego zmniejszenia wydajności. Pułapka jonowa jest zwykle wypełniona helem do ciśnienia około 1 mtorr. Zderzenia z helem tłumią energię kinetyczną jonów i służą do szybkiego kurczenia się trajektorii w kierunku środka pułapki jonowej, umożliwiając wychwytywanie wtryskiwanych jonów. Uwięzione jony są dalej skupione w kierunku środka pułapki poprzez zastosowanie potencjału oscylacyjnego, zwanego podstawowym rf, nałożonego na elektrodę pierścieniową. Jon będzie stabilnie uwięziony w zależności od wartości masy i ładunku jonu, wielkości pułapki jonowej (r), częstotliwości oscylacyjnej podstawowego rf ( w) i amplitudy napięcia na elektrodzie pierścieniowej ( V). Zależność ruchu jonów od tych parametrów opisuje bezwymiarowy parametr qz, qz = 4ev / mr2w2
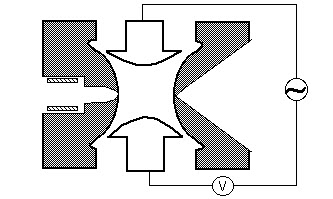
kwadratowy filtr masy składa się z czterech równoległych metalowych prętów ułożonych jak na rysunku poniżej. Dwa przeciwległe pręty mają zastosowany potencjał (U+Vcos(wt)), a pozostałe dwa pręty mają potencjał- (U+Vcos (wt)), gdzie U jest napięciem stałym, a Vcos (WT) jest napięciem przemiennym. Zastosowane napięcia wpływają na trajektorię jonów poruszających się po torze lotu pośrodku między czterema prętami. Dla podanych napięć dc i ac tylko jony o określonym stosunku masy do ładunku przechodzą przez filtr kwadrupolowy, a wszystkie inne jony są wyrzucane z ich pierwotnej drogi. Widmo masowe uzyskuje się przez monitorowanie jonów przechodzących przez filtr kwadrupolowy, gdy napięcia na prętach są zróżnicowane. Istnieją dwie metody: zmienne w i utrzymujące stałą U I V lub zmienne U I V (U/V) ustalone dla stałej w.
kwadratowy filtr masy składa się z czterech równoległych metalowych prętów ułożonych jak na rysunku poniżej. Dwa przeciwległe pręty ha
tandemowa Spektrometria Mas:
tandemowa Spektrometria Mas, zwykle określana jako MS/MS, obejmuje użycie 2 lub więcej analizatorów masy. Jest często używany do analizy poszczególnych składników w mieszaninie. Technika ta dodaje specyficzności danej analizie. Chociaż tandemowa spektrometria MAS może być odnoszona do MS / MS, MS/MS/MS itp…, w niniejszej prezentacji opiszę tylko MS/MS.
podstawową ideą MS/MS jest wybór m/z danego jonu powstałego w źródle jonów i poddanie go fragmentacji, zwykle przez zderzenie z gazem obojętnym (np. Argon). Jony produktu są następnie wykrywane. Jest to potężny sposób potwierdzania tożsamości niektórych związków i określania struktury nieznanych gatunków. MS / MS to proces, który obejmuje 3 etapy: jonizację, dobór masy, analizę masy.
MS / MS może być wykonywane na instrumentach takich jak potrójny kwadrupol (QQQ), pułapka jonowa, Czas lotu, transformata Fouriera itp… Potrójny kwadrupol jest najczęściej stosowanym spektrometrem masowym dla MS / MS, być może ze względu na koszt i łatwość użycia między innymi.
metody separacji dla sprzęgania ze specyfikacją masową
- GC-MS: Mieszaniny próbek są bezpośrednio odparowywane i wprowadzane do połączonych kolumn z topioną krzemionką. Składniki mieszaniny są rozdzielane na podstawie różnicy powinowactwa z fazą wiązaną. Oddzielone związki opuszczają kolumnę i wchodzą do układu próżniowego spektrometru masowego. Cząsteczki próbki są zjonizowane (EI lub CI) i przyspieszane do prekalibrowanego analizatora masy (np. Q, Pułapka jonowa, TOF, FTMS itp…). Rejestrowane są czasy retencji, masy cząsteczkowe i wzorce fragmentacji. Jednym z najważniejszych rozważań GC / MS jest to, że próbka(próbki) musi być nie termolabilna, co oznacza stabilność termiczną.
- LC-MS: w przypadku związków, które są niestabilne termicznie, rozważa się metodę LC/MS. Separacja opiera się na różnicy powinowactwa próbek z fazą stacjonarną i fazą ruchomą. np. hydrofobowość w przypadku chromatografii RP.
- CZE-MS: ta metoda opiera się na różnicach mobilności elektroforetycznej próbek, gdy kolumna stopionej krzemionki znajduje się pod różnicą potencjałów między stroną wtrysku a stroną detektora.
- CIEF-MS: jest to wariant CZE. Opiera się na dyfrerencji w punktach izoelektrycznych analitów.