Ce tutoriel traite des aspects de base de la spectrométrie de masse qui vous seront utiles pour décider des techniques et des mesures appropriées pour vos échantillons de recherche.
- Qu’est-ce que la spectrométrie de masse?
- Le Spectromètre de masse
- Méthodes d’Ionisation
- Ionisation par Impact Électronique
- Ionisation chimique
- Désorption Laser Assistée par Matrice
- Ionisation Chimique à Pression Atmosphérique
- Ionisation Électrospray
- Analyse des ions
- Spectrométrie de masse en tandem
- Méthodes de séparation pour le couplage avec la Spécification de masse
Qu’est-ce que la spectrométrie de masse?
La spectrométrie de masse est une technique analytique qui implique l’étude en phase gazeuse de molécules ionisées dans le but d’un ou plusieurs des éléments suivants:
- Détermination du poids moléculaire
- Caractérisation structurale
- Etude de réactivité en phase gazeuse
- Analyse qualitative et quantitative des composants d’un mélange.
La spectrométrie de masse consiste essentiellement à peser des ions en phase gazeuse. L’instrument utilisé pourrait être considéré comme un équilibre sophistiqué qui détermine avec une grande précision les masses d’atomes et de molécules individuels. Selon les propriétés chimiques et physiques des échantillons, différentes techniques d’ionisation peuvent être utilisées. L’un des principaux facteurs dans le choix de la technique d’ionisation à utiliser est la thermolabilité. Pour les échantillons qui ne sont pas themolabile et relativement volatils, l’ionisation telle que l’Impact électronique et / ou l’Ionisation chimique peut être utilisée efficacement. Pour les échantillons thermolabiles tels que les peptides, les protéines et autres échantillons d’intérêt biologique, des techniques d’ionisation douce sont à envisager. Parmi les techniques d’ionisation douce les plus utilisées figurent l’Électrospray (ESI) et la Désorption Laser assistée par Matrice (MALDI). Le nom donné à une technique de spécification de masse particulière indique généralement la méthode d’ionisation utilisée.
Les masses atomiques et moléculaires sont attribuées par rapport à la masse de l’isotope du carbone, 12C, dont le poids atomique est défini exactement comme 12. La masse réelle de 12C est de 12 daltons, avec un dalton égal à 1.661 10-24 g. La masse d’une molécule ou d’un ion peut être présentée en daltons (Da) ou en kilodaltons (kDa).
Le spectromètre de masse
La spectrométrie de masse utilise un instrument appelé spectromètre de masse. Les principaux composants d’un spectromètre de masse sont:
- Système d’entrée (LC, GC, sonde directe, etc…)
- Source d’ions (EI, CI, ESI, APCI, MALDI, etc…)
- Analyseur de masse (Quadripôle, TOF, Piège À Ions, Secteur Magnétique)
- Détecteur (Multiplicateur d’Électrons, Plaques Micro-Canaux MCPs)
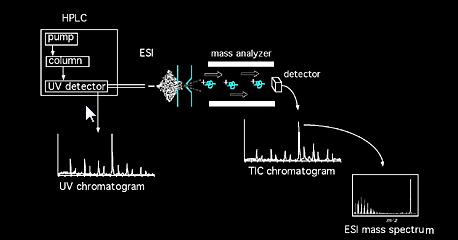
Les échantillons peuvent être introduits directement dans le spectromètre de masse par sonde à solides, ou dans le cas de mélanges, par l’intermédiaire d’un dispositif de chromatographie (par example chromatographie en phase gazeuse, chromatographie en phase liquide, électrophorèse capillaire, etc…). Une fois dans la source, les molécules d’échantillon sont soumises à une ionisation. Les ions formés dans la source (ions moléculaires et fragments) acquièrent une certaine énergie cinétique et quittent la source. Un analyseur calibré analyse ensuite les ions qui passent en fonction de leur rapport masse/charge. Différents types d’analyseur(s) peuvent être utilisés, Magnétique, Quadrulpolaire, Piège à ions, Transformée de Fourier, Temps de vol, etc…Le faisceau d’ions sortant de l’ensemble analyseur est alors détecté et le signal est enregistré. Les acronymes de méthode d’ionisation courants incluent:
- EI = Impact Électronique;
- CI = Ionisation chimique;
- SIMS = Spécification De Masse d’Ions Secondaires;
- FAB = Bombardement Rapide d’Atomes;
- LDMS = Spécification de Masse de Désorption Laser;
- PDMS = Spécification de Masse de Désorption Plasma;
- TS = Thermospray;
- AS= Aerospray;
- ESMS = Spécification de masse d’Électrospray.
Les acronymes d’analyseur de masse courants incluent:
- EB = Électrostatique-magnétique;
- IT = piège à ions;
- Q = Quadripôle;
- TOF = Temps de vol.
Méthodes d’ionisation
La sélection de la méthode d’ionisation appropriée pour l’analyse de votre échantillon est extrêmement importante. Bien que nous puissions vous proposer des suggestions, il est de votre responsabilité de comprendre et de sélectionner la ou les méthodes appropriées pour vos composés de recherche.
- Ionisation EI par Impact électronique
- Ionisation chimique CI
- Ionisation Chimique par Ions négatifs
- Techniques d’Ionisation par Électrospray
- Désorption par Laser assistée par matrice (non offerte dans notre établissement, mais disponible ici sur le campus)
- Ionisation Chimique à Pression atmosphérique APCI
Ionisation par impact électronique
M + e – (70eV) —– > M+. La méthode d’ionisation +2e-
EI convient aux composés non thermolabiles. La volatilité de l’échantillon est requise. Les molécules d’échantillon à l’état de vapeur sont bombardées par des électrons en mouvement rapide, d’énergie conventionnelle de 70 eV. Il en résulte la formation d’ions. Un électron de l’énergie orbitale la plus élevée est délogé et, par conséquent, des ions moléculaires se forment. Certains de ces ions moléculaires se décomposent et des ions fragments se forment. La fragmentation d’un ion donné est due à l’excès d’énergie qu’il nécessite au sein de l’ionisation. Les ions fragments peuvent être des électrons impairs ou même des électrons. Les ions moléculaires formés lors de l’ionisation par impact électronique sont des ions électroniques impairs. Les ions de fragments d’électrons impairs sont formés par clivage direct (p.ex. clivage direct d’une liaison C-C). Même les ions fragments d’électrons sont souvent formés par réarrangement (par exemple, transfert de protons). L’échantillon peut être introduit dans la source EI via un dispositif de chromatographie en phase gazeuse, par example dans le cas de mélanges, ou directement via un dispositif de sonde de solides. Les quantités nécessaires à une expérience sont généralement inférieures à un microgramme de matière.
Les spectres de masse EI, dans la plupart des cas, contiennent des pics d’ions fragmentaires intenses et des pics d’ions moléculaires beaucoup moins intenses. Lorsque le pic d’ions moléculaires n’est pas observé dans le spectre de masse, l’ionisation chimique peut être utilisée afin d’obtenir des informations sur les ions moléculaires. Une règle utile pour déterminer si un ion est un ion moléculaire est la règle de l’azote.
Règle de l’azote: Comme indiqué ci-dessus, les ions moléculaires formés lors de l’ionisation EI sont des ions électroniques impairs. Si leur rapport masse/charge observé est impair, la molécule étudiée contient un nombre impair d’atomes d’azote. Si ce rapport masse / charge est un nombre pair, cette molécule ne contient pas ou même d’atomes d’azote.
Ionisation chimique
Pour les chimistes organiques, l’ionisation chimique (CI) est une technique particulièrement utile lorsqu’aucun ion moléculaire n’est observé dans le spectre de masse EI, et également dans le cas de la confirmation du rapport masse / charge de l’ion moléculaire. La technique d’ionisation chimique utilise pratiquement le même dispositif de source d’ions que dans l’impact électronique, sauf que CI utilise une source d’ions serrée et un gaz réactif. Le gaz réactif (par exemple l’ammoniac) est d’abord soumis à un impact électronique. Les ions d’échantillon sont formés par l’interaction des ions de gaz réactif et des molécules d’échantillon. Ce phénomène est appelé réactions ion-molécule. Les molécules de gaz réactif sont présentes dans un rapport d’environ 100:1 par rapport aux molécules d’échantillon. Des ions positifs et des ions négatifs sont formés dans le processus CI. En fonction de la configuration de l’instrument (tensions de source, détecteur, etc…) seuls les ions positifs ou seuls les ions négatifs sont enregistrés.
Dans l’IC, des réactions de molécules d’ions se produisent entre des molécules de gaz réactif ionisées (G) et des molécules neutres d’analyte volatiles (M) pour produire des ions d’analyte. Les ions pseudo-moléculaires MH+ (mode ionique positif) ou – (mode ionique négatif) sont souvent observés. Contrairement aux ions moléculaires obtenus dans la méthode EI, la détection MH + et – se produit avec un rendement élevé et moins d’ions fragmentés sont observés.
Mode d’ions positifs:
GH ++ M —— > MH + + G
Mode d’ions négatifs:
– +M ——> – + G
Ces réactions simples de transfert de protons sont de véritables processus Acide-Base en phase gazeuse au sens de Bronsted-Lowrey. Une source d’ions « serrée » (pression = 0,1-2 torr) est utilisée pour maximiser les collisions, ce qui augmente la sensibilité. Pour avoir lieu, ces réactions de molécules d’ions doivent être exothermiques. Le transfert de protons est l’un des processus simples observés en CI positif :
RH ++ M —– > MH ++ R
L’un des paramètres décisifs de cette réaction est l’affinité des protons. Pour que la réaction se produise, l’affinité protonique de la molécule M doit être supérieure à celle de la molécule de gaz. Les principaux gaz réactifs utilisés dans l’IC sont: l’ammoniac, le méthane et l’isobutane. Les ions réactifs prédominants formés sont donnés dans les mécanismes illustrés ci-dessous. Le choix du gaz réactif affecte l’étendue de la fragmentation de l’ion quasi moléculaire.
Méthane (ionisation chimique à ions positifs):
- CH4 + e —–> CH4+. + 2e ——> CH3+ + H.
- CH4+. + CH4 —–> CH5+ +CH3.
- CH4+. + CH4 —–> C2H5+ + H2 + H.
Isobutane (positive ion chemical ionization):
- i-C4H10 + e —–> i-C4H10+. + 2e
- i-C4H10+. + i-C4H10 ——> i-C4H9+ + C4H9 +H2
Ammonia (positive ion chemical ionization):
- NH3 + e —–> NH3+. + 2e
- NH3+. + NH3 ——> NH4+ + NH2.
- NH4++ NH3 ———> N2H7+
En ionisation chimique en mode d’ions positifs au méthane, les pics d’échantillons pertinents observés sont MH+, + et +; mais principalement MH+. Cela correspond aux masses M+1, M+29 et M+41.
En ionisation chimique en mode isobutane à ions positifs, le pic principal observé est MH+.
En ionisation chimique en mode ammoniac positif, les principaux pics observés sont MH+ et +. Si plus d’un site de protonation est présent, des adduits NH3 supplémentaires peuvent être observés correspondant à +. Cela correspond aux masses M+1, M+18 et M+35.
Dans certains cas, des dimères protonés ou d’autres adduits peuvent être observés; une perte de H2O suivie d’une protonation ou de la formation d’ions adduits est observée pour certaines classes de composés. Si le spectre que vous observez ne semble pas montrer les ions adduits appropriés, ou montre une fragmentation étendue, méfiez-vous lorsque vous essayez d’interpréter les résultats. Il existe une abondance de données disponibles dans la littérature traitant des mécanismes d’ionisation chimique applicables à des classes spécifiques de composés.
Deux facteurs déterminent le choix du gaz réactif à utiliser:
- Affinité protonique PA
- Transfert d’énergie
NH3 (ammoniac) est le gaz réactif le plus utilisé dans l’IC en raison du faible transfert d’énergie du NH4+ comparé au CH5 + par exemple. Avec le NH3 comme gaz réactif, on observe généralement MH+ et MNH4+ (différence de 17 unités de masse).
Ionisation chimique par ions négatifs
Trois mécanismes peuvent être soulignés:
- Réaction de capture d’électrons due à la réalisation d’électrons « thermalisés » à faible énergie et à déplacement lent qui peuvent être transférés plus efficacement aux molécules d’échantillon.
- Transfert d’électrons à partir d’un gaz réactif ionisé (par exemple NH2 – peut transférer un électron vers une molécule ayant une plus grande affinité électronique que NH2).
- Les ions de gaz réactifs participent aux réactions d’IC vraies (par exemple, abstraction de protons, selon les acidités relatives).
Les ions moléculaires observés dans les spectres de masse d’ionisation chimique à ions négatifs sont généralement M- ou -.
Méthode d’ionisation par électrospray
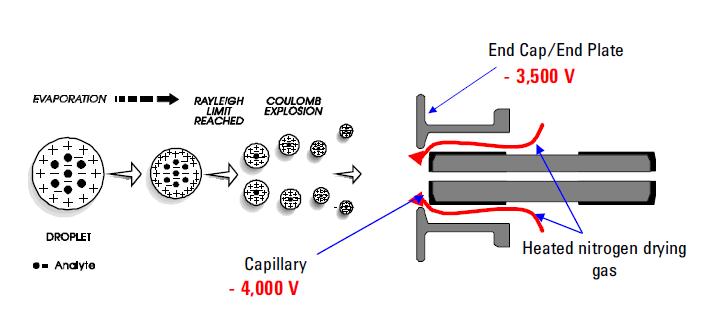
Parmi les techniques d’ionisation par pulvérisation les plus utilisées, on trouve l’ionisation par pulvérisation électrospray (ESI). Cette technique continue d’être la méthode de choix pour analyser les produits chimiques thermolabiles. Ses capacités sont bien documentées. Il utilise une contrainte électrique entre la sortie de la sonde ESI (par exemple capillaire) et la contre-électrode, située à quelques millimètres de la sonde. Le processus entraîne la génération de gouttelettes fortement chargées directement à partir de la solution infusée. Multiplier et / ou désorber les molécules d’analyte chargées individuellement à partir des gouttelettes pulvérisées et échantillonnées à travers le reste du spectromètre de masse. L’ESI se distingue par sa capacité à produire des ions moléculaires chargés en plusieurs fois à partir d’une grande variété de polymères tels que des fragments de protéines et d’ADN; il permet également la détection sensible d’espèces polaires de faible poids moléculaire chargées individuellement telles que les médicaments et les métabolites de médicaments. La formation d’ions positifs ou négatifs (selon le signe du champ électrique appliqué) se produit avec un rendement élevé. En mode ionique positif, des molécules d’analyte d’adduits protonés et/ou alcalins sont généralement observées dans les spectres de masse. En mode ions négatifs, des pics de fonctionnement correspondent à des molécules d’analyte déprotonées sont observés. L’ESI est décrite comme une technique d’ionisation très « douce » où le gaz du bain environnant a un effet modérateur sur les énergies internes et de translation des ions désorbés.
Avantages de l’ESI:
- Processus d’ionisation douce permettant d’observer des ions moléculaires intacts
- ESI permet la production d’ions chargés en plusieurs fois. Il en résulte la capacité d’analyser des espèces de très haut poids moléculaire à l’aide des analyseurs de masse les plus disponibles (par exemple, les quadrupoles).
- ESI est un procédé à pression atmosphérique. Cela le rend facile à utiliser et facile à interfacer avec les techniques de séparation HPLC et CE.
Désorption Laser Assistée par Matrice (MALDI)
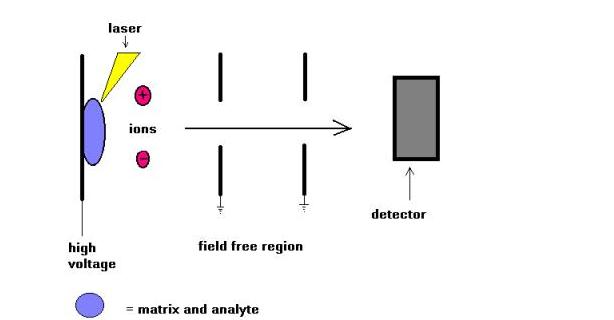
La technique de spectrométrie de masse par désorption Laser assistée par matrice (MALDI) a été introduite par Karas et Hillkamp en 1988 pour l’ionisation de peptides et de protéines. Bientôt, cette technique a pu analyser d’autres types de biomolécules, tels que des oligosaccharides, des glycolipides, des nucléotides et des polymères synthétiques. Dans cette technique, les échantillons sont cocristallisés avec une substance absorbant les UV appelée matrice. Par exemple pour les protéines, la matrice de choix est souvent l’acide sinapinique. Un rayonnement de 337 nm provenant d’un laser à azote est le plus couramment utilisé. Le laser aide à introduire de l’énergie dans le système moléculaire de manière à empêcher la décomposition thermique.
MALDI est souvent utilisé avec les spectromètres de masse à temps de vol (TOF) en raison de la nature pulsante de la technique et de la capacité de plage de masse. Des poids moléculaires pouvant atteindre quelques centaines de daltons ont pu être mesurés. La comparaison des techniques d’ionisation MALDI et ESI a été tentée au cours des dernières années. À mon avis, ces deux techniques ne sont pas compétitives mais complémentaires. Pour n’en nommer que quelques-uns, pour les espèces de haut poids moléculaire, MALDI conduit à la formation d’ions moléculaires chargés individuellement tandis que ESI permet la formation d’ions moléculaires chargés en plusieurs fois.
Considérations pratiques:
- Le rapport molaire final échantillon/matrice est d’environ 1/5000.
- La concentration finale de l’échantillon est de 1 à 10 pmol / ul
- Notre expérience avec MALDI indique une plage dynamique de 100 fmol / ul à quelques centaines pmol / ul
- MALDI est une technique d’ionisation relativement robuste qui tolère l’utilisation de sels et de tensioactifs et de tampons. Bien qu’il soit préférable de les enlever pour de meilleures performances.
Normes peptidiques et protéiques pour MALDI:
- Angiotensine II (humaine) MW: 1046,2
- Substance P (humaine) MW: 1347,7
- Insuline (bovine) MW: 5733,6
- Cytochrome c (équine) MW: 12 360.1
- RNase A (bovine) MW: 13,682.2
- Apo-Myoglobine (équine) MW: 16,951.5
- Trypsinogène (bovin) MW: 23,980.9
Ionisation Chimique à Pression Atmosphérique
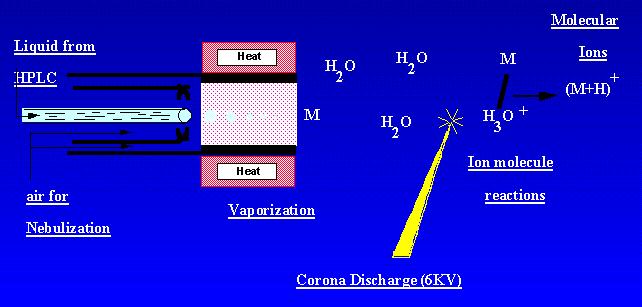
L’APCI est une technique qui crée des ions à pression atmosphérique. Une solution d’échantillon s’écoule à travers un tube chauffé où elle est volatilisée dans un brouillard et pulvérisée dans une décharge corona à l’aide de la nébulisation à l’azote. Les échantillons moléculaires sont ionisés par des réactions de molécules d’ions à partir des ions de décharge de la couronne ambiante. Les ions sont produits dans la décharge et extraits dans le spectromètre de masse. APCI est le mieux adapté aux échantillons semi-volatils relativement polaires. Un spectre de masse APCI contient généralement l’ion quasi-moléculaire, – ou +.
Analyse des ions
Il est possible d’utiliser plusieurs paramètres physiques différents pour obtenir une séparation de masse. Les types courants d’analyseurs de masse sont discutés ci-dessous. Chacun a des avantages et des inconvénients. Dans nos installations, nous disposons actuellement de spectromètres de masse Quadripolaires, à piège à ions et à temps de vol amd (TOF).
Spectromètre de Masse à Secteur magnétique
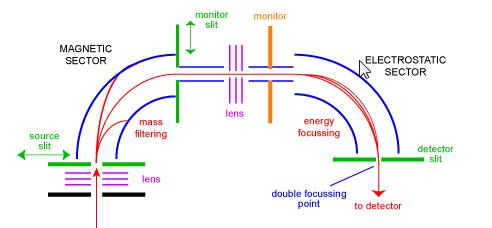
Le spectromètre de masse sectoriel était l’un des types d’analyseurs de masse les plus courants et probablement le plus familier au scientifique de tous les jours. Dans les années 1950, les premiers spectromètres de masse commerciaux étaient des instruments sectoriels. Ils consistent en une combinaison d’un grand dispositif électromagnétique et d’une sorte de dispositif de focalisation électrostatique. Différents fabricants utilisent des géométries différentes. La figure 1 montre un schéma d’un instrument à double focalisation à géométrie » BE » standard. La configuration BE est également appelée spectromètre de masse à secteur à géométrie inverse, c’est-à-dire un instrument à double secteur composé d’un secteur magnétique suivi d’un secteur électrostatique.
Les ions pénètrent dans l’instrument à partir de la source (en bas à gauche) où ils sont initialement focalisés. Ils pénètrent dans le secteur magnétique par la fente de source où ils sont déviés selon la règle de gauche. Les ions de masse supérieure sont moins déviés que les ions de masse inférieure. Le balayage de l’aimant permet de focaliser des ions de masses différentes sur la fente du moniteur. A ce stade, les ions n’ont été séparés que par leurs masses. Pour obtenir un spectre de bonne résolution où tous les ions de même m/z apparaissent coïncidant comme un pic dans le spectre, les ions doivent être filtrés par leurs énergies cinétiques. Après une autre étape de focalisation, les ions pénètrent dans le secteur électrostatique où les ions du même m/z voient leurs distributions d’énergie corrigées et sont focalisés au point de focalisation double sur la fente du détecteur.
Les instruments sectoriels ont connu d’énormes succès commerciaux dans les années 1950 et 1960, car ils étaient le seul moyen pratique d’obtenir des données à haute résolution. Au cours des 20 dernières années environ, avec la baisse des prix des FTMS et le développement d’alternatives à haute résolution (par exemple Q-Tof), les instruments du secteur sont en déclin.
Spectrométrie de masse à temps de vol (TOF-MS)
Un spectromètre de masse à temps de vol utilise les différences de temps de transit à travers une région de dérive pour séparer des ions de masses différentes. Il fonctionne en mode pulsé, de sorte que les ions doivent être produits ou extraits en impulsions. Un champ électrique accélère tous les ions dans une région de dérive sans champ avec une énergie cinétique de qV, où q est la charge d’ions et V est la tension appliquée. Étant donné que l’énergie cinétique des ions est de 0,5mv2, les ions plus légers ont une vitesse plus élevée que les ions plus lourds et atteignent le détecteur à la fin de la région de dérive plus tôt.
Théorie:
- K.E. = qV
- 1/2 mv2 = qV
- v =(2qV/m)1/2
Le temps de transit (t) à travers le tube de dérive est L / v où L est la longueur du tube de dérive
- t = L /(2V / m / q)1/2
Schéma d’un TOF-MS linéaire
Ce schéma montre l’ablation d’ions d’un échantillon solide avec un laser pulsé. Le réflectron est une série d’anneaux ou de grilles qui agissent comme un miroir ionique. Ce miroir compense la propagation des énergies cinétiques des ions lorsqu’ils entrent dans la région de dérive et améliore la résolution de l’instrument. La sortie d’un détecteur d’ions est affichée sur un oscilloscope en fonction du temps pour produire le spectre de masse.
Piège à ions
Les ions créés par impact électronique (EI), électrospray (ESI) ou ionisation par désorption laser assistée par matrice (MALDI) sont focalisés à l’aide d’un système de lentilles électrostatiques dans le piège à ions. Une grille d’ions électrostatique pulse ouverte (-V) et fermée (+V) pour injecter des ions dans le piège à ions. La pulsation de la porte ionique différencie les pièges ioniques des instruments à « faisceaux » tels que les quadripôles où les ions pénètrent continuellement dans l’analyseur de masse. Le temps pendant lequel les ions sont admis dans le piège, appelé « durée d’ionisation », est réglé pour maximiser le signal tout en minimisant les effets de charge d’espace. La charge d’espace résulte d’un trop grand nombre d’ions dans le piège qui provoquent une distorsion des champs électriques entraînant une réduction globale des performances. Le piège à ions est généralement rempli d’hélium à une pression d’environ 1 mtorr. Les collisions avec l’hélium amortissent l’énergie cinétique des ions et servent à contracter rapidement des trajectoires vers le centre du piège à ions, ce qui permet de piéger les ions injectés. Les ions piégés sont davantage focalisés vers le centre du piège grâce à l’utilisation d’un potentiel oscillant, appelé rf fondamental, appliqué à l’électrode annulaire. Un ion sera piégé de manière stable en fonction des valeurs de la masse et de la charge de l’ion, de la taille du piège à ions (r), de la fréquence d’oscillation de la rf fondamentale (w) et de l’amplitude de la tension sur l’électrode annulaire (V). La dépendance du mouvement ionique sur ces paramètres est décrite par le paramètre sans dimension qz, qz = 4eV/ mr2w2
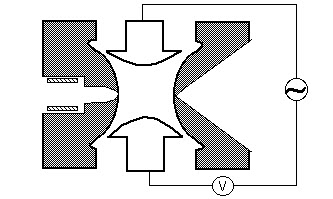
Quadripôle
Un filtre de masse quadripolaire est constitué de quatre tiges métalliques parallèles disposées comme sur la figure ci-dessous. Deux tiges opposées ont un potentiel appliqué de (U + Vcos (wt)) et les deux autres tiges ont un potentiel de – (U + Vcos (wt)), où U est une tension continue et Vcos (wt) est une tension alternative. Les tensions appliquées affectent la trajectoire des ions se déplaçant le long de la trajectoire de vol centrée entre les quatre tiges. Pour des tensions continues et alternatives données, seuls les ions d’un certain rapport masse / charge traversent le filtre quadripolaire et tous les autres ions sont expulsés de leur chemin d’origine. Un spectre de masse est obtenu en surveillant les ions traversant le filtre quadripolaire au fur et à mesure que les tensions sur les bâtonnets sont variées. Il existe deux méthodes: faire varier w et maintenir U et V constants, ou faire varier U et V (U / V) fixés pour une w constante.
Un filtre de masse quadripolaire est constitué de quatre tiges métalliques parallèles disposées comme sur la figure ci-dessous. Deux tiges opposées ha
Spectrométrie de masse en tandem:
La spectrométrie de masse en tandem, généralement appelée MS / MS, implique l’utilisation de 2 analyseurs de masse ou plus. Il est souvent utilisé pour analyser les composants individuels d’un mélange. Cette technique ajoute de la spécificité à une analyse donnée. Bien que la spectrométrie de masse en tandem puisse être référée à MS / MS, MS / MS / MS, etc…, dans cette présentation, je vais décrire uniquement MS / MS.
L’idée de base de MS / MS est une sélection d’un m / z d’un ion donné formé dans la source d’ions, et soumet cet ion à une fragmentation, généralement par collision avec un gaz inerte (par exemple. Argon). Les ions produits sont alors détectés. C’est un moyen puissant de confirmer l’identité de certains composés et de déterminer la structure d’espèces inconnues. Ainsi, MS / MS est un processus qui implique 3 étapes: ionisation, sélection de masse, analyse de masse.
MS / MS pourrait être effectué sur des instruments tels que le triple quadripôle (QQQ), le piège à ions, le temps de vol, la transformée de fourier, etc… Le triple quadripôle est le spectromètre de masse le plus fréquemment utilisé pour MS / MS, peut-être en raison du coût et de la facilité d’utilisation, entre autres facteurs.
Méthodes de séparation pour le couplage avec la spécification de masse
- GC-MS: Les mélanges d’échantillons sont directement vaporisés et pénètrent dans des colonnes de silice fondue liée. Les composants du mélange sont séparés en fonction de leur différence d’affinité avec la phase liée. Les composés séparés sortent de la colonne et entrent dans le système de vide du spectromètre de masse. Les molécules d’échantillon sont ionisées (EI ou CI) et accélérées dans un analyseur de masse précalibré (par exemple Q, Piège à ions, TOF, FTMS, etc…). Les temps de rétention, les masses moléculaires et les schémas de fragmentation sont enregistrés. L’une des considérations les plus importantes de la GC / MS est que le ou les échantillons doivent être non thermolables, ce qui signifie qu’ils sont thermiquement stables.
- LC-MS: Pour les composés thermiquement instables, la méthode LC / MS est considérée. La séparation est basée sur la différence d’affinité des échantillons avec la phase stationnaire et la phase mobile. par exemple, hydrophobicité en cas de chromatographie RP.
- CZE-MS : Cette méthode est basée sur les diffrences de mobilité électrophorétique des échantillons lorsque la colonne de silice fondue est sous une différence de potentiel entre côté injection et côté détecteur.
- CIEF-MS : Il s’agit d’une variante de CZE. Il est basé sur les différences dans les points isoélectriques des analytes.