fysiologisten ominaisuuksien, kuten entsyymiaktiivisuuden, kvantitatiiviset mittarit ilmaistaan usein aktiivisuusyksikköinä milligrammaproteiinia kohti. Vaikka proteiinipitoisuuden mittaamiseksi on kehitetty lukuisia määrityksiä, kuten amido Black (1), Biuret (2), Bikinikoniinihappo (3) ja Coomassie Blue (4,5) – kolorimetriset määritykset, Lowry-määritystä (6) tai sen muunnoksia (7,8) käytetään yleisemmin kuin muita määrityksiä (9). Lowry-määritysmenetelmä on yksinkertainen, herkkä ja tarkka, ja se on viitatuin (10) menetelmä proteiinin kvantitatiivisessa määrityksessä.
monenlaiset yhdisteet, jotka reagoivat Folin-Ciocalteu-fenolin (Foliinin) reagenssin (11) kanssa, voivat aiheuttaa häiriöitä Lowry-ja modifioiduissa Lowry-proteiinimäärityksissä. Onneksi korjaus asianmukaisen nollan avulla riittää useimmille yhdisteille (6,7) paitsi lipideille (12), pesuaineille (13) ja värillisille aineille (14). Modifioidulla Lowry-määrityksellä (15; tässä asiakirjassa U-1988-määritys, 16) voitettiin vaikeudet proteiinien määrityksessä lipidien ja pesuaineiden läsnä ollessa (käytetään rasvakudoksen, myeliinin ja luurankolihasten liukoisuuteen). Punaviinin valkuaispitoisuuden määrittämiseen (14,17,18) liittyvät värihäiriöt voitettiin käyttämällä laajaa kromatografiaa. Edellä mainittu lähestymistapa on hankala eikä kovin käytännöllinen suurten näytteiden käsittelyyn. Mikään tunnetuista proteiinimäärityksistä ei soveltunut proteiinien mittaamiseen värillisistä biologisista näytteistä esim., värilliset hedelmät ja vihannekset, punaviini, pigmentoidut mikrobit ja märehtijöiden sappi.
kehittäessämme U-2012-määritystä sen edeltäjistä U-1988: sta ja Lowry-määrityksestä on saavutettu kolme merkittävää etua: I) helppous reagenssiformulaatioiden stabiilisuuden ansiosta, ii) proteiinin mittaaminen sekä värittömistä että värillisistä biologisista näytteistä herkkyyttä vaarantamatta ja iii) proteiinien määritys hyvin pieninä pitoisuuksina. Tätä uutta määritystä sovelletaan proteiinin kvantitatiiviseen määrittämiseen sekä värittömistä että värillisistä biologisista näytehomogenaateista, mukaan lukien runsaasti lipidejä sisältävät (esim.avokado) ja vaikeasti homogenoitavat.
- materiaalit ja menetelmät
- biologiset näytteet – punajuuri -, mustikka-ja punaviini
- kemialliset reagenssit
- parannukset U-1988-määritykseen
- U-2012-määritys
- Värihäiriöiden estimointi U-2012-määrityksessä
- standardikäyriä ja sen parametreja
- valkuaispitoisuuden laskeminen homogenaateissa
- tulokset ja keskustelu
- U-1988-määrityksen parannukset
- valkuaisarvio värillisissä biologisissa näytteissä
- Proteiiniuute
- eliminoidaan häiritsevät aineet
- Standardikäyrät ja niiden parametrit
- värillisen homogenaatin proteiinipitoisuus
- kiitokset
- kilpailevat intressit
- lisätiedot
materiaalit ja menetelmät
punajuuri-ja mustikkahomogenaatit valmistettiin lisäaineiston mukaisesti. Punaviini ei vaatinut proteiinin uuttamista ennen U-2012-määritystä.
kemialliset reagenssit
kaikki kemialliset reagenssit natriumhypokloriittia ja perkloorihappoa (PCA) lukuun ottamatta saatiin Sigmasta tai Sigma-Aldrichista (St Louis, MO. YHDYSVALLAT). Natriumhypokloriitti oli peräisin Acros Organicista, New Jerseystä, Yhdysvalloista. PCA saatiin BDH: lta (Englanti).
parannukset U-1988-määritykseen
siirtyminen karbonaatista fosfaattipuskuriin pH: ssa 12,0 paransi reagenssin stabiilisuutta ja lisäsi hieman herkkyyttä. Asetonitriili otettiin käyttöön pesuaineiden aiheuttamien kuplien välttämiseksi. NaOH korvasi Koh: n, jotta se ei saostuisi proteiinimäärityksessä. Lisäksi tehokkuutta parannettiin yhdistämällä Lowry-reagenssin eri komponentit yhdeksi reagenssisekoitukseksi.
U-2012-määritys
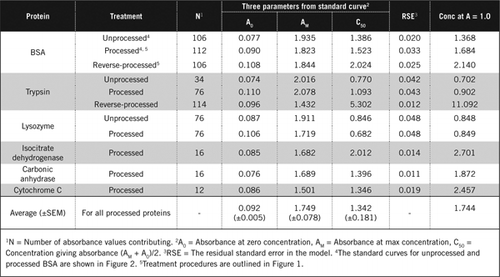
Täydelliset tiedot U-2012-määrityksestä on esitetty täydentävässä aineistossa. Kuviossa 1 lyhyesti tiivistetyssä pöytäkirjassa kuvataan punaviinin ja punajuuri-ja mustikkahomogenaattien käsittelyä sekä u-1988-määrityksen parannuksia. U-2012-määritystä käytettiin käsittelemättömistä, käsitellyistä ja käänteisesti käsitellyistä proteiineista (H2O2-käsittely, jota seuraa TCA-tai PCA-saostus). Määritykset tehtiin BSA: lle, hiilihappoanhydraasille, sytokromi C: lle, isositraattidehydrogenaasille, lysotsyymille ja trypsiinille standardikäyrien kehittämiseksi ja värillisissä biologisissa näytteissä. Biologisten näytteiden proteiinit määritettiin kalibroimalla sopiviin standardikäyriin.
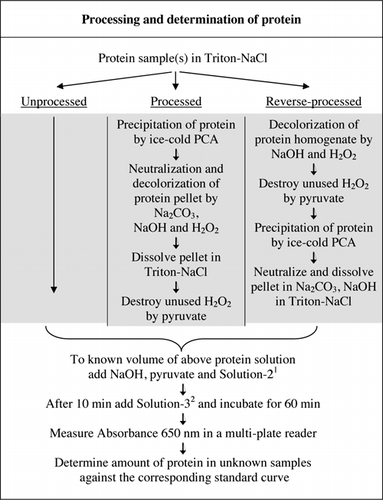
1solution-2 sisälsi kuparisulfaattia (CuSO4.5H2O), Na-K-tartraattia, SDS: ää ja asetonitriiliä 100 mM: n fosfaattipuskurissa (pH 12.0). 2for Solution-3, Folin-Ciocalteun fenolireagenssi laimennettiin 1:1 deionisoidulla vedellä juuri ennen käyttöä.
Värihäiriöiden estimointi U-2012-määrityksessä
värihäiriöt määritettiin vertaamalla käsiteltyjen ja käsittelemättömien punajuuri -, mustikka-ja punaviininäytteiden imukykyä sekä Folin-reagenssilla että ilman sitä Kuvan 1 mukaisesti. Suhdetta käytettiin interferenssin laajuuden määrittämiseen, jossa Abs1 on käsittelemättömien näytteiden absorbanssi Foliinin reagenssilla; Abs2 on käsittelemättömien näytteiden absorbanssi ilman Foliinin reagenssia; Abs3 on käsiteltyjen näytteiden absorbanssi Foliinin reagenssilla; ja Abs4 absorboi käsiteltyjä näytteitä ilman Folinin reagenssia.
standardikäyriä ja sen parametreja
oheisen lisäaineiston Reseptit-osiossa kuvattuja ratkaisuja-1B ja 1c käytettiin standardikäyrien kehittämiseen. BSA: n pitoisuus ja vastaavat absorbanssiarvot piirrettiin X-Y-hajontagrammilla. Tämän kaavion muoto (kuva 2) osoittaa kyllästyvän vasteen suuremmilla pitoisuuksilla ja hyvin rajallisen alkuperäisen lineaarisen vasteen. Tämä oli aiemmin ilmoitettu ensisijainen käyrä (20). Aluksi tämä mallinnettiin käyttäen eksponentiaalista muotoa (19), mutta myöhemmät tutkimukset osoittivat, että suorakulmainen Hyperbeli antoi paremman kohdistuksen vasteeseen, erityisesti pienemmillä pitoisuuksilla. Tämä jälkimmäinen muoto on nyt standardoitu ja seuraavaa kolmen parametrin yhtälöä käytettiin kuvaamaan absorbanssin ja proteiinin konsentraatiosuhdetta:

Conc = proteiinipitoisuus,
a = absorbanssi konsentraatiossa
A0= absorbanssi nollakonsentraatiossa,
AM= absorbanssi maksimipitoisuudessa,
C50 = konsentraatio, joka antaa absorbanssin
(AM + A0)/2.
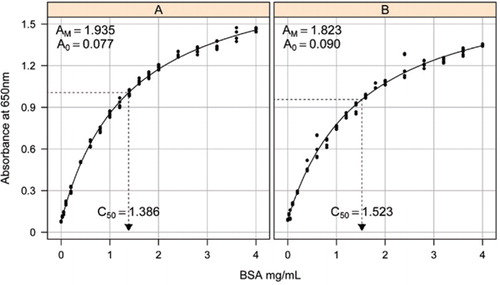
absorbanssin (650 nm) ja BSA-proteiinin pitoisuuksien välinen standardikäyrä Estimoitiin kolmella teknisellä rinnakkaisnäytteellä a) käsittelemättömän proteiinin määritykseen (parametrit A0= 0, 077, Am=1, 935, C50=1).386) ja B) käsitellyn proteiinin määritys (parametrit A0=0, 90, Am=1, 823, C50=1, 523). Havaitulle datalle asennettiin suorakulmainen hyperbelimalli (yhtälö). Parametri-estimaatit tunnistetaan käyrissä, joissa on X-akselin C50-estimaatti (musta pisteviiva).
parametri A0 oli kokeellisesti määritetty, kun taas AM ja C50 arvioitiin käyttäen Microsoft Excelin Toolbox add-in ratkaisija toiminto. Koeparametrien avulla laskettiin mallinnettu absorbanssi kullakin vakiopitoisuudella (Conc) yhtälön avulla . Ratkaisija oli sitten käsketty minimoimaan jäljellä keskihajonta mitattu ja mallinnettu absorbanssi standardin asettaa säätämällä AM ja C50.
havaitsimme absorbanssin ja konsentraation välisen suhteen osoittavan epälineaarisen käyrän koko pitoisuusalueella, mikä todennäköisesti johtuu valon sironnan komponentista, joka kasvaa proteiinipitoisuuden kasvaessa absorbanssia mitattaessa. Heikko lineaarinen fit Alhainen absorbanssi raportoitiin myös Coakley ja James (20).
valkuaispitoisuuden laskeminen homogenaateissa
Määritykset tehtiin jalostetulle ja käsittelemättömälle punaviinille sekä punajuuri-ja mustikkahomogenaateille. BSA-ja muita proteiininäytteitä käsiteltiin samalla tavalla asianmukaisten standardikäyrien määrittämiseksi A0 -, AM-ja C50-arvojen määrittämiseksi. Näitä parametreja käytettiin sitten muuntamaan näytteen absorbanssi (A) kunkin homogenaatin proteiinipitoisuudeksi käyttäen:

koska yhtälöllä on kyllästyvä muoto, herkkyys pienenee absorbanssin (A) ja siten konsentraation kasvaessa. Proteiinien estimoinneissa olevat virheet voidaan minimoida säätämällä homogenaattien pitoisuuksia määrityksessä niin, että ne eivät ylitä liikaa C50-arvoa.
Homogenaattikonsentraatio muunnettiin sitten kudosproteiinikonsentraatioksi (Kudoskonsentraatio mg / g kudosta) seuraavan kaavan mukaisesti:

jossa Homogenaattikonsentraatio (mg proteiinia / mL) on korjattu mahdollisen prekonsentraation tai laimennuksen osalta määrityksen aikana. Homogenaattiprosentti oli 100 g kudosta homogenoitu kokonaistilavuudeksi 200 mL (meidän tapauksessamme 50%).
erillisessä tutkimuksessa asennettiin suorakulmainen hyperbelimalli käyttäen non linear mixed effects (nlme) – pakettia (21) R (22) (kuva 2). Jokainen laboratoriossa itsenäisesti tehty BSA-ratkaisu mallinnettiin satunnaisvaikutuksena, jossa oli yhteinen A0, mutta erilaiset AM-ja C50-kertoimet. Tämä mallintaa biologisten näytteiden rinnakkaisnäytteiden ja teknisten määritysten rinnakkaisnäytteiden hierarkiaa.
tulokset ja keskustelu
U-1988-määrityksen parannukset
U-1988-ja Lowry-määrityksen rajoitus on karbonaattipohjaisen reagenssin epävakaus. U-1988: ssa karbonaattipuskuri (pH 11,4, 2% = 188,7 mM) korvattiin 40 mM fosfaatilla pH-arvoilla 11,4-12,5. PROTEIINIMÄÄRITYKSEN standardikäyristä, joissa käytettiin BSA: Ta 0, 5 mg: n ja 1, 0 mg: n BSA: ta/ mL, laskettiin ensimmäiset rinteet. Alkurinteet fosfaattipuskureilla pH: ssa 11, 4 ja optimaalisessa pH: ssa 12, 0 olivat 99 x 10-6 ja 197 x 10-6vaste. Karbonaattipuskurin kaltevuus (pH 11,4) oli 162 x10-6.Koska kulmakerroin on suora osoitus määrityksen herkkyydestä, fosfaattipuskuri (pH 12.0) valittiin korvaamaan karbonaattipuskuri, jolloin herkkyys kasvoi 25%.
parempi stabiilisuus saavutettiin nostamalla fosfaattipuskurin pitoisuus 100 mM: iin.saatu fosfaatti/CuSO4/Na-K-tartraattiliuos säilyi huoneenlämmössä kaksi viikkoa, huomattavasti pidempään kuin karbonaatti/CuSO4/Na-K-tartraattiliuos, joka on valmistettava päivittäin ennen proteiinimääritystä. Kaikissa tulevissa kokeissa CuSO4/Na-K-tartraattiliuoksen valmistamiseen käytettiin 100 mM fosfaattia (pH 12.0). Uskomme, että karbonaatin korvaaminen fosfaatilla parantaa U-2012-määrityksen mukavuutta.
pesuaineiden aiheuttamista kuplista tulee suuri virhelähde absorbanssimittauksissa, kun käytetään monikaivolevylukijaa (ei ongelma kuvettien kanssa). Nämä kuplat vähenivät huomattavasti lisäämällä useita polaarisia liuottimia (esim.asetoni, asetonitriili, etanoli ja metanoli). Asetonitriili, näistä liuottimista polaarisin (23) valittiin sen tehokkuuden vuoksi ja se sisällytettiin liuokseen-2 (ks.Kuva 1 kuvateksti ja reseptit-osio Lisämateriaalista).
Fosfaattipuskuri, CuSO4, Na-K-tartraatti, SDS ja asetonitriili voidaan lisätä yksittäin, eikä niiden yhteenlaskujärjestys vaikuta syntyvään absorbanssiin. Esiseostetun liuoksen käyttö lisää kuitenkin käyttömukavuutta, erityisesti silloin, kun on määrä määrittää suuri määrä näytteitä. Tämän vuoksi ryhmitimme nämä testiseoksen komponentit liuokseen-2 (Kuva 1). Tällaista esiseostettua liuosta ei voitu käyttää alkuperäisessä Lowry-määrityksessä (6) karbonaattiliuoksen epästabiilisuuden vuoksi. Yritys sisällyttää Solution-3 Solution-2: een johti sinisen värin kehityksen dramaattiseen vähenemiseen, eikä sitä harkittu sen enempää.
valkuaisarvio värillisissä biologisissa näytteissä
Proteiiniuute
punajuuren ja mustikan proteiinit uutettiin Triton X-100-NaCl-liuoksessa miedolla homogenoinnilla. Tällaiset homogenaatit säilyttävät entsyymiaktiivisuutensa (15). Uuttamista ei vaadittu punaviinin osalta.
eliminoidaan häiritsevät aineet
värillisten näytteiden osalta on tarpeen poistaa häiriö, joka johtuu näytteen luontaisesta väristä ja muista proteiinireagenssien kanssa reagoivista ei-proteiineista ennen kolorimetristä proteiinimääritystä. U-2012: n uutuus on värinpoistoprotokollan laatiminen, joka on yhteensopiva kolorimetrisen proteiinimäärityksen kanssa.
värillisten pigmenttien värjäämistä natriumhypokloriitin tai H2O2: n avulla ja proteiinien valikoivaa saostamista PCA: n tai TCA: n avulla harkittiin häiritsevien aineiden poistamiseksi. Natriumhypokloriitti, H2O2, TCA ja PCA arvioitiin niiden yhteensopivuuden U-2012-määrityksen kanssa, jossa testiproteiinina käytettiin BSA: ta. Natriumhypokloriitin ja H2O2: n välillä vain H2O2 oli yhteensopiva, koska hypokloriitin läsnä ollessa muodostui saostumaa. TCA: n tai PCA: n saostamat proteiinit voidaan määrittää U-2012: lla pelletin jäännöshapon riittävän neutraloinnin jälkeen. On raportoitu PCA: n paremmuudesta TCA: han verrattuna proteiinin saostumisessa (24,25). Sen sijaan vertailevassa arvioinnissamme havaittiin samanlaisia C50-arvoja PCA: lle (1.395) ja TCA: lle (1.400). Suosimme PCA: ta, koska se on helposti saatavilla valmiiksi valmistettuna ratkaisuna (70% v/v) ja siksi helposti laimennettuna vaadittuun vahvuuteen. TCA on hygroskooppinen kiinteä aine, jota on vaikea punnita tarkasti vaihtelevan vesipitoisuutensa vuoksi.
on kaksi mahdollista tapaa yhdistää PCA ja H2O2. ”Käsitellyille ”proteiineille PCA-hoitoa seurasi H2O2-hoito ja” käänteisesti käsitellyille ” proteiineille H2O2-hoito edelsi PCA-saostumista. ”Käsitellyn” proteiinin käytön etuja olivat useiden häiritsevien aineiden poistaminen supernatantista ja proteolyyttisten entsyymien mahdollinen inaktivoituminen näytteiden valmistuksen aikana. Tämä vahvistettiin määrittämällä käsitelty ja käänteisesti käsitelty trypsiini ja BSA (KS.Taulukko 1). Värillisten biologisten näytteiden todellisen proteiinipitoisuuden määrittämiseen käytettiin vain käsiteltyjä näytteitä
värillisten näytteiden sekä PCA-että H2O2-käsittelyt olivat tarpeen interferenssin poistamiseksi U-2012-määrityksessä. Pelkästään värillisten näytteiden happosaostuma ei poistanut häiriöitä kokonaan. Kaikilla värillisillä näytteillä supernatanttiin heitettiin jonkin verran väriä, mutta myös pelletit olivat värillisiä. Väri poistui pelleteistä H2O2-käsittelyllä. Emäksisiä olosuhteita vaadittiin sekä H2O2: n (26) tehokkaaseen värinpoistoon että folinin reagenssin värinkehitykseen, jotta valkuaispitoisuudet mitattaisiin oikein. Vaikka sekä NaOH että KOH pystyivät tuottamaan vaaditun emäksisyyden, vain NaOH oli yhteensopiva U-2012-määrityksen kanssa. Koh: n läsnä ollessa muodostui sakka. Pelleteissä PCA neutraloitiin Na2CO3: lla ja NaOH: lla (27). Määrityksen aikana lisättiin lisää NaOH: ta; optimoitu tilavuus oli 50-70 µL (rutiininomaisesti käytettiin 60 µL); KS.Kuva 1.
punajuuri, mustikka ja punaviini dekoloroitiin siten, että 15 µL 30% H2O2 otettiin 0,5 ja 2 h 50°C: n lämpötilassa ja huoneenlämmössä. Voimakkaampien värillisten näytteiden käsittelyyn käytettiin kahtakymmentä mikrolitraa, joiden H2O2-pitoisuus oli 30% 1 tunnin ajan 50°C: ssa. H2O2: n proteiineihin sitoutuneiden sokereiden kaltaisten aineiden hapettuminen 50°C: ssa vaikuttaa kriittiseltä, koska huoneenlämmössä prosessointi yliarvioi valkuaispitoisuuden. Punajuuren tapauksessa 50°C: n käsittely alensi näennäisen proteiinin estimaatin 14 prosenttiin käsittelemättömästä proteiinista, kun taas huoneenlämmössä tapahtuva käsittely vain puolitti tämän arvion.
vetyperoksidikäsittelyn jälkeen tehdyistä kolorimetrisistä määrityksistä kävi ilmi, että osaa H2O2: sta ei käytetty värinpoistossa. Tällaisessa näytteessä Lowry-määrityksen päätyväri tuhoutui osittain. Jäljelle jäänyt vetyperoksidi oli siksi hävitettävä ennen proteiinimääritystä. U-2012-määrityksessä on kaksi H2O2: n lähdettä; lisätty H2O2 värinpoistoa varten ja H2O2 esiintyy vierasaineena Triton X-100: ssa (0,22%, Tuotetiedot: Triton X-100, www.sigmaaldrich.com). vetyperoksidia hajoaa yleisesti katalaasientsyymin vaikutuksesta. Proteiinimäärityksen korkea pH kuitenkin inaktivoisi tunnetut katalaasit. Katalaasin lisääminen johtaisi myös lisäproteiinin lisäämiseen. Valitsimme H2O2: n kemiallisen tuhoamisen pyruvaatin (28) avulla. Pyruvaatti-H2O2-vuorovaikutusyhtälön kemia on vakiintunut (28,29). Pyruvaatti tuhoaa H2O2: n huoneenlämmössä seuraavalla reaktiolla:

pellettisuspensiossa oleva H2O2-jäännös tuhottiin käsittelemällä 0,9 M pyruvaatilla (1,5 x H2O2-pitoisuus) 0,5 tunnin ajan huoneenlämmössä. Triton X-100: ssa olevan kontaminoivan H2O2: n torjumiseksi proteiinimääritykseen lisättiin myös ylimääräistä pyruvaattia (Kuva 1). Pyruvaatin lisääminen antoi alhaisemman absorbanssin ei-proteiiniselle nollalle . Ehdotamme, että Triton X-100: n sisältämä peroksidi reagoi Asetonitriilin kanssa liuoksessa-2, jolloin imukyky on hieman korkeampi.
värillisiin biologisiin näytteisiin liittyviä väri-häiriöitä ei voida ottaa huomioon yksinkertaisesti suorittamalla proteiinimääritys Foliinin reagenssin puuttuessa. Lasketut suhdeluvut (Abs1-Abs2)/(Abs3-Abs4) osoittivat, että näyteväristä johtuvia häiriöitä oli eniten punaviinillä ( = 40) ja vähemmän mustikalla ( = 6) ja punajuurella ( = 2). Tämä häiriö johti poikkeuksellisen korkeisiin arvioihin todellisista proteiinipitoisuuksista; esimerkiksi proteiinipitoisuus käyttämällä jalostamattomia ja jalostettuja punajuurihomogenaatteja (20,21 ja 2,89 mg proteiinia / g kudosta). Värihäiriön lisäksi punaviini sekä punajuuren ja mustikan homogenaatit sisältävät todennäköisesti aineita, jotka reagoivat Folinin reagenssin kanssa U-2012-määrityksessä (e.g., pienet peptidit ja kompleksisokerit). Nämä poistuivat saostamalla selektiivisesti proteiineja jääkylmällä PCA: lla lopullisena pitoisuutena 5% (kuva 1).
Standardikäyrät ja niiden parametrit
käsittelemätöntä ja käsiteltyä BSA: ta koskevat Standardikäyrät on esitetty kuvassa 2. Johdetut parametrit (A0, AM ja C50) on lueteltu myös taulukossa 1 BSA: n ja muiden proteiinien osalta.
tulokset osoittavat, että mallin jäännöskeskivirhe on pieni (0,012-0,048), mikä osoittaa datan paremman istuvuuden suorakulmaiseen hyperbelitrendiin. Vertailtaessa tietoa eri proteiinien välillä ja niiden käsittelyä muuttujat muunnettiin absorbanssikonsentraatioksi = 1, 0 650 nm: ssä (taulukon 1 oikea sarake).
nämä tulokset osoittavat, että valkuaisainehäviö (verrattuna käsittelemättömään proteiiniin) prosessoiduissa näytteissä oli pienempi kuin käänteisesti käsitellyissä näytteissä. Tämä menetys oli selvempi trypsiinin tapauksessa, ja se voidaan selittää sen autokatalyyttisen aktiivisuuden perusteella käänteisprosessoinnin aikana. Suosittelemme, että ”prosessoitua” protokollaa (lisäaineistoa) noudatetaan vain sellaisten biologisten näytteiden osalta, jotka todennäköisesti sisältävät proteolyyttisiä entsyymejä.
alkuperäisessä Lowry-määrityksessä (6) ja sen modifioidussa versiossa U-1988 (15) proteiinin kvantitatiivisessa määrityksessä käytettiin vain standardikäyrän lineaarista osaa, joka saatiin määrittämällä absorbanssi proteiinin määrää vastaan. U-2012-määrityksessä tietoja käytetään tehokkaammin sovittamalla materiaalit ja menetelmät-osiossa kuvattu suorakulmainen hyperbeliyhtälö Coakleyn ja Jamesin (20) mukaisesti.
värillisen homogenaatin proteiinipitoisuus
valkuaispitoisuudet tuntemattomissa näytteissä laskettiin yhtälöllä ja prosessoidun BSA-standardin ja taulukossa 1 lueteltujen kaikkien prosessoitujen proteiinien keskiarvon perusteella. Jälkimmäinen on lähempänä todellista arviota biologisista näytteistä, jotka sisältävät valkuaisaineseoksen. Arvioimme mustikan ja punajuuren proteiinimäärät suhteessa punaviiniin noin 60-ja 230-kertaisiksi (Taulukko 2).
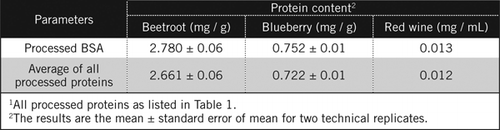
kuten BSA, punaviini ja 50% homogenaatit punajuuri ja mustikka jalostettiin PCA saostamalla ja dekolorisoimalla H2O2 (Kuva 1). Tässä vaiheessa biologisia näytteitä tiivistettiin punaviinille 40 kertaa ja punajuurelle ja mustikalle 4 kertaa. Samoin myös BSA: Ta (2 mg/mL) konsentroitiin 4-8 mg/mL. Värillisten näytteiden absorbanssia, joka oli lähellä absorbanssia C50: lle (käsittelemättömälle BSA: lle), käytettiin proteiinipitoisuuden laskemiseen yhtälön ja kuvaamalla tavalla .
yhteenvetona voidaan todeta, että U-2012-määrityksessä on käytetty stabiileja reagensseja, parannettu herkkyyttä (jopa värittömille biologisille näytteille) ja voitettu värillisten biologisten näytteiden aiheuttama värihäiriö. U-2012-määritystä ei rajoiteta proteiinipitoisuuden ja absorbanssin välisen vasteen lineaariseen osaan, ja se tehostaa epälineaarisen alueen tietojen käyttöä suorakulmaisen hyperbolisen käyrämallin avulla, joka on asennettu standardeihin käyttäen yksinkertaisia menetelmiä Microsoft Excelissä.
kiitokset
kirjoittajat tunnustavat tutkimuksen, tieteen ja teknologian Uuden-Seelannin taloudellisen tuen säätiön (C06x0809).
kilpailevat intressit
kirjoittajat ilmoittavat, ettei kilpailevia intressejä ole.
lisätiedot
tämän asiakirjan liitteenä olevat lisätiedot löytyvät lehden verkkosivuilta osoitteesta: www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R. S. ja P. L. Pedersen. 1985. Mikrogramman määrien määrittäminen proteiinista milligramman lipiditasojen läsnä ollessa amido black 10B. Anal. Biochem. 150:97–104.Crossref, Medline, CAS, Google Scholar
- 2. Gornall, A. G., C. J. Bardawill ja M. M. David. 1949. Seerumin proteiinien määrittäminen biuret-reaktiolla. J. Biol. Kemiaa. 177:751–766.Medline, CAS, Google Scholar
- 3. Smith, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, et al.. 1985. Proteiinin mittaus bikinikoniinihapolla. Anaali. Biochem. 150:76–85.Crossref, Medline, CAS, Google Scholar
- 4. Bradford, M. M. 1976. Nopea ja herkkä menetelmä proteiinin mikrogrammamäärien kvantifioimiseksi käyttäen proteiinivärin sitoutumisperiaatetta. Anaali. Biochem. 72:248–254.Crossref, Medline, CAS, Google Scholar
- 5. Zor, T. ja Z. Selinger. 1996. Bradford protein assayn linearisointi lisää sen herkkyyttä: teoreettiset ja kokeelliset tutkimukset. Anaali. Biochem. 236:302–308.Crossref, Medline, CAS, Google Scholar
- 6. Lowry, O. H., N. J. Rosbrough, A. L. Farr ja R. J. Randall. 1951. Proteiinin mittaus Folin fenolireagenssilla. J. Biol. Kemiaa. 193:265–275.Medline, CAS, Google Scholar
- 7. Peterson, G. L. 1979. Review of the Folin phenol protein quantification method of Lowry, Rosebrough, Farr and Randall. Anaali. Biochem. 100:201–220.Crossref, Medline, CAS, Google Scholar
- 8. SAPA, CV, RL. Lundablad ja N. C. Price. 1999. Kolorimetriset proteiinimääritystekniikat. Bioteknologiaa. Appl. Biochem. 29:99–108.Medline, CAS, Google Scholar
- 9. Okutucu, B., A. Dınçer, Ö. Habib ja F. Zıhnıoglu. 2007. Viiden menetelmän vertailu plasman kokonaisproteiinipitoisuuden määrittämiseksi. J. Biochem. Biofyysejä. Menetelmät 70: 709-711.Crossref, Medline, CAS, Google Scholar
- 10. Kresge, N., R. D. Simoni ja R. L. Hill. 2005. The most highly siteered paper in publishing history: protein determination by Oliver H. Lowry. J. Biol. Kemiaa. 25:280.Google Scholar
- 11. Everette, J. D., Q. M. Bryant, A. M. Green, Y. A. Abbey, G. W. Wangila ja R. B. Walker. 2010. Perusteellinen tutkimus eri yhdisteluokkien reaktiivisuudesta Folin-Ciocalteu-reagenssiin nähden. J. Agric. Ruokakemikaalia. 58:8139–8144.Crossref, Medline, CAS, Google Scholar
- 12. Eichberg, J. ja L. C. Mokrasch. 1969. Hapettuneiden lipidien aiheuttama häiriö proteiinin määrityksessä Lowry-menetelmällä. Anaali. Biochem. 30:386–390.Crossref, Medline, CAS, Google Scholar
- 13. Dulley, J. R. ja P. A. surevat. 1975. Yksinkertainen tekniikka pesuaineiden aiheuttamien häiriöiden poistamiseksi Lowry-proteiininmääritysmenetelmässä. Anaali. Biochem. 64:136–141.Crossref, Medline, CAS, Google Scholar
- 14. Brillouet, J.-M., M.-P. Belleville ja M. Moutouet. 1991. Mahdolliset proteiini-polysakkaridikompleksit punaviineissä. On. J. Enol. Vitic. 42:150–152.CAS, Google Scholar
- 15. Upreti, G. C., R. A. Ratcliff ja P. C. Riches. 1988. Proteiinin estimointi suuria lipidipitoisuuksia sisältävissä kudoksissa: modifications to Lowry method of protein determination. Anaali. Biochem. 168:421–427.Crossref, Medline, CAS, Google Scholar
- 16. Upreti, G. C., C. Davis ja J. Oliver. 1991. Preparation of representative homogenates of biological kudokset: effect of salt on protein extraction. Anaali. Biochem. 198:298–301.Crossref, Medline, CAS, Google Scholar
- 17. Smith, M. R., M. H. Penner, S. E. Bennett ja A. T. Bakalinsky. 2011. Kvantitatiivinen kolorimetrinen määritys punaviiniin Pinot Noir levitetylle Kokonaisproteiinille. J. Agric. Ruokakemikaalia. 59:6871–6876.Crossref, Medline, CAS, Google Scholar
- 18. Wigand, P., S. Tenzer, H. Schild ja H. Decker. 2009. Punaviinin proteiinikoostumuksen analyysi verrattuna Rosé-ja valkoviineihin Elektroforeesillä ja Korkeapainenestekromatografisella massaspektrometrialla (HPLC-MS). J. Agric. Ruokakemikaalia. 57:4328–4333.Crossref, Medline, CAS, Google Scholar
- 19. Upreti, G. C., Y. Wang, A. Sharrock, N. Feisst, M. Davy ja B. Jordan. 2009. Stabiili ja herkkä proteiinimääritys (U-2009 modified assay) värillisille biologisille näytteille. ComBiol., Uuden-Seelannin Lopullinen Ohjelma Joulukuu 2009. Canterburyn yliopisto, Christchurch, Uusi-Seelanti.Google Scholar
- 20. Coakley, W. T. ja C. J. James. 1978. Yksinkertainen lineaarinen muunnos Folin-Lowry-proteiinin kalibrointikäyrälle arvoon 1.0 mg/mL. Anaali. Biochem. 85:90–97.Crossref, Medline, CAS, Google Scholar
- 21. Pinheiro, J. C. ja D. M. Bates. 2000. Sekaefektimallit S-ja S-PLUS -, tilasto-ja Laskentasarjoissa. Springer-Verlag, New York, NY.Google Scholar
- 22. R Development Core Team. 2009. R: kieli ja ympäristö tilastollista tietojenkäsittelyä varten. R Foundation for Statistical Computing, Wien, Itävalta, ISBN 3-900051-07-0, URL http://www.R-project.org.Google Scholar
- 23. Khachik, F., G. R. Beecher, J. T. Vanderslice ja G. Vako. 1988. Nestekromatografiset artefaktit ja huippuvääristymä: näyte-liuotin-vuorovaikutus karotenoidien erottamisessa. Anaali. Kemiaa. 60:807–811.Crossref, Medline, CAS, Google Scholar
- 24. Cernik, AAA 1970. Etyleenidiamiinietikkahapon kanssa kelatoidun lyijyn määrittäminen verestä sen jälkeen, kun proteiini on saostettu perkloorihapolla. Brit. J. Teollisuus Med. 27:40–42.Medline, CAS, Google Scholar
- 25. Moughan, P. J., A. J. Darragh, W. C. Smith ja C. A. Butts. 1990. Perklooriset ja trikloorietikkahapot proteiinin saostajina endogeenisessa ileaalisessa digestassa rotasta. J. Sci. Food Agric. 52:13–21.Crossref, CAS, Google Scholar
- 26. Galbács, Z. M. ja L. J. Csányi. 1983. Alkali-indusoitu hajoaminen vetyperoksidia. J. Kemi. Soc. Dalton Trans. 11:2353–2357.Crossref, Google Scholar
- 27. Scopes, RK 1988. Protein Purification: Principles and Practice, Second Ed. Springer-Verlag New York Inc., New York, NY.Google Scholar
- 28. Upreti, G. C., K. Jensen, R. Munday, D. M. Duganzich, R. Vishwanath ja J. F. Smith. 1998. Tutkimukset aromaattisten aminohappojen oksidaasiaktiivisuudesta ram-siittiöissä: pyruvaatin rooli antioksidanttina. Animia. Reprod. Sci. 51:275–287.Crossref, Medline, CAS, Google Scholar
- 29. Holleman, Maf 1904. Notice sur l ’action de l’ eau oxygenee sur les acids α-cetoniques et sur les dicetones 1.2. Recl. Trav. Chim. Pays-Bas Belg. 23:169–172.Crossref, CAS, Google Scholar