medidas Quantitativas de traços fisiológicos, tais como a atividade da enzima normalmente são expressos como unidades de atividade por miligrama de proteína. Embora tenham sido desenvolvidos numerosos ensaios para medir o teor de proteínas, incluindo os ensaios colorimétricos de Amido Negro (1), Biuret (2), Ácido Bicinchonínico (3) e Coomassie Blue (4,5), o ensaio de Lowry (6) ou as suas modificações (7,8) são mais frequentemente utilizados do que outros ensaios (9). O ensaio de Lowry é simples, sensível e preciso, sendo o procedimento mais citado (10) para a determinação quantitativa de proteínas.
uma grande variedade de compostos que reagem com o reagente Folin-Ciocalteu fenol (reagente de Folin) (11) são uma fonte de potencial interferência em ensaios de proteínas Lowry e de proteínas Lowry modificadas. Felizmente, correções através de um branco apropriado é suficiente para a maioria dos compostos (6,7) exceto lípidos (12), detergentes (13) e substâncias coloridas (14). As dificuldades na determinação das proteínas na presença de lípidos e detergentes (utilizados na solubilização dos tecidos adiposos, mielina e músculos esqueléticos) foram superadas pelo ensaio modificado de Lowry (15; referido neste artigo como ensaio U-1988, 16). A interferência da cor na determinação do teor de proteínas no vinho tinto (14,17,18) foi superada através da utilização de cromatografia extensiva. A abordagem acima é complicada e pouco prática para o manuseamento de um grande número de amostras. Nenhum dos testes de proteínas conhecidos foram adequados para medir proteínas em amostras biológicas coloridas., frutas e vegetais coloridos, vinho tinto, micróbios pigmentados e bílis ruminante.
o desenvolvimento da Nossa U-2012 ensaio dos seus antecessores, o U-1988 e o Lowry ensaio obteve três grandes vantagens (i) a conveniência através da estabilidade das formulações de reagentes, (ii) a medida de proteína em ambos os incolor e colorido amostras biológicas, sem comprometer a sensibilidade, e (iii) análise de proteínas em concentrações muito baixas. Este novo ensaio será aplicável à determinação quantitativa de proteínas em homogeneizados de amostra biológica incolor e colorida, incluindo aqueles ricos em lípidos (por exemplo, abacate) e aqueles difíceis de homogeneizar.
- Materiais e métodos
- amostras Biológicas – beterraba, mirtilo e vinho tinto
- reagentes químicos
- melhorias no ensaio de u-1988
- a estimativa das interferências de cores no doseamento de U-2012
- a curva-padrão e os seus parâmetros
- o cálculo do teor de proteínas nos homogeneizados
- Results and discussion
- Improvements in the U-1988 assay
- a estimativa de proteínas em amostras biológicas coloridas
- a extracção de proteínas
- eliminar substâncias interferentes
- agradecimentos
- interesses concorrentes
- dados Suplementares
Materiais e métodos
amostras Biológicas – beterraba, mirtilo e vinho tinto
Beterraba e mirtilo homogenates foram preparados como descrito no Material Suplementar. O vinho tinto não necessitava de extração de proteínas antes do ensaio U-2012.
reagentes químicos
todos os reagentes químicos, excepto hipoclorito de sódio e ácido perclórico (PCA), foram obtidos a partir de Sigma ou Sigma-Aldrich (St.Louis, MO. AMERICA). O hipoclorito de sódio era da Acros Organic, New Jersey, EUA. O PCA foi obtido da BDH (Inglaterra).
melhorias no ensaio de u-1988
mudança do tampão de carbonato para fosfato a pH 12, 0 melhorou a estabilidade do reagente e deu um pequeno aumento na sensibilidade. O acetonitrilo foi introduzido para evitar bolhas induzidas por detergentes. NaOH substituiu KOH para evitar a precipitação no ensaio de proteínas. Além disso, a eficiência foi melhorada através da combinação de vários componentes do reagente de Lowry em uma mistura de reagente.O Material suplementar fornece dados completos sobre o doseamento de U-2012 . O protocolo, resumido brevemente na Figura 1, descreve a transformação do vinho tinto e os homogeneizados da beterraba e do mirtilo, e inclui as melhorias do ensaio U-1988. O ensaio U-2012 foi utilizado para proteínas processadas não processadas, processadas e processadas inversamente (tratamento H2O2 seguido de precipitação AP ou APC). Foram efectuados ensaios em ASC, anidrase carbónica, citocromo C, desidrogenase isocitrato, lisozima e tripsina para o desenvolvimento de curvas padrão e em amostras biológicas coloridas. A determinação das proteínas nas amostras biológicas foi realizada calibrando-as para curvas padrão apropriadas.
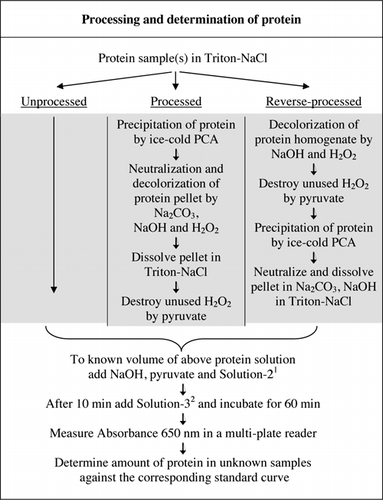
1Solution-2 contém sulfato de cobre (CuSO4.5H2O), Na-K-tartarato, SDS e acetonitrilo em tampão fosfato de 100 mM (pH 12.0). 2for Solução-3, o reagente de fenol de Folin-Ciocalteu foi diluído 1:1 com água desionizada imediatamente antes da utilização.
a estimativa das interferências de cores no doseamento de U-2012
foi determinada comparando a absorvência das amostras de beterraba processada e não processada, mirtilo e vinho tinto, tanto com como sem a utilização do reagente de Folin, como descrito na Figura 1. O rácio foi utilizado para determinar a extensão da interferência, em que o Abs1 é absorvência de amostras não processadas com o reagente de Folin; o Abs2 é absorvência de amostras não processadas sem o reagente de Folin; o Abs3 é absorvância de amostras processadas com o reagente de Folin; e o Abs4 é absorvância de amostras processadas sem reagente de Folin.
a curva-padrão e os seus parâmetros
solução-1b e 1C descritos na secção receitas do material suplementar anexado foram utilizados para o desenvolvimento das curvas-padrão. A concentração de BSA e os valores de absorvância correspondentes foram desenhados utilizando um grafo de dispersão X-Y. A forma deste gráfico (Figura 2) mostra uma resposta saturada em concentrações mais elevadas, com uma resposta linear inicial muito limitada. Esta foi uma forma de curva preferida relatada anteriormente (20). Inicialmente isso foi modelado usando uma forma exponencial (19), mas estudos posteriores mostraram que uma hipérbole retangular deu um melhor alinhamento com a resposta, particularmente em concentrações mais baixas. Esta última forma foi agora padronizada e a seguinte equação de três parâmetros foi usada para descrever a relação de concentração de absorvância-proteína:

Conc = concentração de Proteína,
A = Absorvância a Conc,
A0 = Absorbância zero de concentração,
AM= Absorvância máxima concentração,
C50= Concentração dando absorvância
(AM + A0)/2.
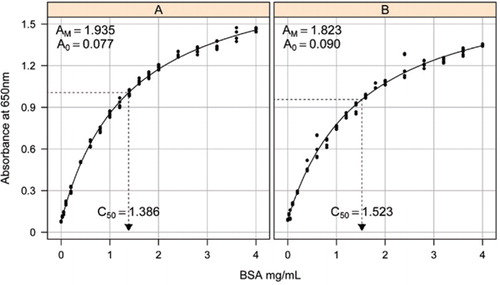
a estimativa da curva-padrão entre a absorvância (650 nm) e as concentrações de proteína BSA foi feita em três replicados técnicos a) o doseamento de proteínas não transformadas (parâmetros A0= 0, 077, Am=1,935, C50=1.386), e B) o doseamento de proteínas processadas (parâmetros A0=0, 90, Am=1, 823, C50=1, 523). Um modelo de hipérbole retangular (equação ) foi adaptado aos dados observados. As estimativas dos parâmetros são identificadas nas curvas com a estimativa de C50 no eixo dos x (linha preta pontilhada).
o parâmetro A0 foi determinado experimentalmente, enquanto AM e C50 foram estimados usando a função add-in Solver do Microsoft Excel. Foi utilizado um conjunto experimental de parâmetros para calcular a absorvância modelada em cada uma das concentrações padrão (Conc) utilizando a equação . A solução foi então comandada para minimizar o desvio-padrão residual entre a absorvância medida e modelada para o conjunto-padrão, ajustando AM e C50.
observamos que a relação entre a absorvância e a concentração apresenta uma curva não linear ao longo de toda a gama de concentrações, o que provavelmente se deve a um componente de espalhamento de luz que aumenta à medida que a concentração de proteínas aumenta ao medir a absorvância. Um mau ajuste linear em baixa absorvância também foi relatado por Coakley e James (20).
o cálculo do teor de proteínas nos homogeneizados
foi efectuado em vinhos tintos transformados e não transformados e em homogeneizados de beterraba e de mirtilo. As BSA e outras amostras de proteínas foram tratadas de forma idêntica para curvas padrão adequadas para determinar os valores A0, AM e C50. Estes parâmetros foram então utilizados para converter a absorvância da amostra (a) em concentração proteica em cada homogeneizado, utilizando:

como a equação tem uma forma saturada, a sensibilidade reduz-se à medida que a absorvância (A), e, portanto, a concentração, aumenta. Os erros nas estimativas de proteínas podem ser minimizados ajustando as concentrações de homogeneizados no ensaio de modo a não excederem excessivamente o valor C50.
O Homogeneizado Conc valor foi convertido em tecido concentração de proteína (Tecido Concentração em mg/g de tecido), utilizando a seguinte fórmula:

onde Homogeneizado Conc (em mg de proteína/mL) foi corrigido para qualquer pré-concentração ou diluição durante o ensaio. A percentagem de homogeneização foi de 100 g de tecido homogeneizado até um volume total de 200 mL (no nosso caso 50%).
num estudo separado, foi instalado um modelo de hipérbole rectangular utilizando o pacote de efeitos mistos não lineares (nlme) (21) em R (22) (Figura 2). Cada solução BSA, feita independentemente no laboratório, foi modelada como um efeito aleatório, com um comum A0 mas diferentes coeficientes AM e C50. Isto modela a hierarquia dos replicados da amostra biológica e dos replicados do ensaio técnico.
Results and discussion
Improvements in the U-1988 assay
The limitation of the U-1988 and The Lowry assay is the instability of the carbonate-based reagent. O tampão carbonatado (pH 11,4 a 2% = 188,7 mM) em u-1988 foi substituído por fosfato de 40 mM com valores de pH compreendidos entre 11,4 e 12,5. Calcularam-se inclinações iniciais a partir das curvas padrão do ensaio proteico utilizando ASC a 0, 5 mg ASC/ mL e 1, 0 mg ASC / mL. As encostas iniciais com tampões de fosfato a pH 11,4 e com pH óptimo de 12,0 foram, respectivamente, 99 x10-6 e 197 x10-6. O declive para o tampão de carbonato (pH 11.4) foi 162 x10-6.Dado que o valor do declive é uma indicação directa da sensibilidade do doseamento, tampão fosfato (pH 12.0) foi escolhido para substituir o tampão carbonatado, dando um aumento de 25% na sensibilidade.A solução de fosfato/CuSO4/Na-K-tartarato resultante foi estável à temperatura ambiente durante duas semanas, consideravelmente mais longa do que a solução de carbonato/CuSO4/Na-K-tartarato, que deve ser preparada diariamente antes do ensaio com proteínas. Para todas as experiências futuras, foi utilizado fosfato de 100 mM (pH 12,0) para preparar a solução CuSO4/Na-K-tartarato. Acreditamos que a substituição do carbonato pelo fosfato irá melhorar a conveniência do ensaio U-2012.
as bolhas induzidas pelo detergente tornam-se uma importante fonte de erro nas medições da absorvância quando se utiliza um leitor de placas multi-alvéolos (não é um problema com as cuvetas). Estas bolhas foram consideravelmente reduzidas pela adição de uma série de solventes polares (por exemplo, acetona, acetonitrilo, etanol e metanol). O acetonitrilo, o mais polarizado destes solventes (23) foi escolhido pela sua eficácia e incluído na solução-2 (ver Figura 1 legenda e secção de receitas do material suplementar).
tampão fosfato, CuSO4, Na-K-tartarato, SDS e acetonitrilo podem ser adicionados individualmente e a ordem da sua adição não afecta a absorvância resultante. No entanto, a utilização de uma solução pré-misturada aumenta ainda mais a conveniência, especialmente quando um grande número de amostras devem ser ensaiadas. Assim, agrupamos estes componentes da mistura de ensaio em Solução-2 (Figura 1). Esta solução pré-misturada não foi viável para o ensaio original de Lowry (6) devido à instabilidade da solução de carbonato. A tentativa de incluir a solução-3 na solução-2 resultou em redução dramática no desenvolvimento da cor Azul e não foi considerada mais.
a estimativa de proteínas em amostras biológicas coloridas
a extracção de proteínas
as proteínas de beterraba e mirtilo foram extraídas na solução Triton X-100-NaCl com homogeneização ligeira. Estes homogeneizados mantêm as suas actividades enzimáticas (15). Esta extração não era necessária para o vinho tinto.
eliminar substâncias interferentes
para amostras coloridas é necessário remover a interferência devido à cor inerente da amostra e outras substâncias não proteicas que reagem com os reagentes proteicos antes do ensaio com a proteína colorimétrica. A novidade do U-2012 é a elaboração de um protocolo de descoloração compatível com um ensaio colorimétrico de proteína.
descoloração de pigmentos coloridos por hipoclorito de sódio ou H2O2 e precipitação selectiva de proteínas por APC ou ACT foram considerados para remoção de substâncias interferentes. O hipoclorito de sódio, o H2O2, o TCA e o PCA foram avaliados quanto à sua compatibilidade com o ensaio U-2012 utilizando a BSA como proteína de ensaio. Entre hipoclorito de sódio e H2O2, apenas H2O2 era compatível como um precipitado foi formado na presença de hipoclorito. As proteínas precipitadas pelo ácido TCA ou PCA podem ser determinadas pelo U-2012 após uma neutralização adequada do ácido residual no sedimento. Foi relatada a superioridade do APC sobre o apt na precipitação de proteínas (24,25). Em contraste, em nossa avaliação comparativa, revelou valores semelhantes de C50 para PCA (1.395) e TCA (1.400). Preferimos o PCA porque está prontamente disponível como uma solução pré-fabricada (70% v/v) e, portanto, facilmente diluída para a resistência necessária. TCA é um sólido higroscópico que é difícil de pesar precisamente devido ao seu teor de água variável.
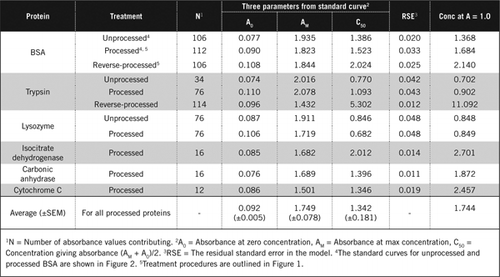
existem duas formas possíveis de combinar PCA e H2O2. Para proteínas” processadas”, o tratamento PCA foi seguido pelo tratamento H2O2 e para proteínas” processadas inversamente”, o tratamento H2O2 precedeu a precipitação de PCA. As vantagens da utilização de proteínas “processadas” foram a remoção de uma série de substâncias interferentes no sobrenadante e a possível inactivação das enzimas proteolíticas durante a preparação da amostra. Este facto foi confirmado pela determinação da tripsina processada e processada reversa e da BSA (ver Quadro 1). Apenas foram utilizadas amostras processadas para determinar o teor real de proteínas de amostras biológicas coloridas
tanto o tratamento PCA como o H2O2 de amostras coloridas foram necessários para a eliminação da interferência no ensaio U-2012. Apenas a precipitação ácida das amostras coloridas não removeu completamente a interferência. Com todas as amostras coloridas, alguma cor foi descartada no sobrenadante, mas as pelotas também foram coloridas. A cor foi eliminada das pastilhas pelo tratamento H2O2. Condições alcalinas foram necessárias para a descoloração eficaz por H2O2 (26) e o desenvolvimento de cor pelo reagente de Folin para garantir que os níveis de proteínas são medidos corretamente. Embora tanto NaOH quanto KOH pudessem fornecer a alcalinidade necessária, apenas NaOH era compatível com o ensaio U-2012. Um precipitado foi formado na presença de KOH. Em pellets, o PCA foi neutralizado usando Na2CO3 e NaOH (27). Adicionou-se NaOH adicional durante o ensaio; o volume optimizado situou-se entre 50 e 70 µL (utilizou-se rotineiramente 60 µL); ver Figura 1.
Beetroot, mirtilo e vinho tinto foram descolorizados com 15 µL de 30% H2O2 tomando 0,5 e 2 h a 50°C e à temperatura ambiente, respectivamente. Vinte microlitros de 30% H2O2 para 1 h a 50°C foi usado para lidar com amostras coloridas mais fortes. A oxidação de substâncias como os açúcares ligados às proteínas por H2O2 a 50°C parece crítica, uma vez que o processamento à temperatura ambiente sobrestima o teor de proteínas. No caso da beterraba sacarina, a transformação a 50°C reduziu a estimativa da proteína aparente para 14% da não transformada, enquanto a transformação à temperatura ambiente apenas reduziu para metade essa estimativa.
foi evidente a partir dos ensaios colorimétricos realizados após o tratamento com peróxido de hidrogénio que alguns H2O2 não foram utilizados na descoloração. Nessa amostra, a cor final do ensaio de Lowry foi parcialmente destruída. Por conseguinte, foi necessário destruir o peróxido de hidrogénio restante antes do ensaio com proteínas. Há duas fontes de H2O2 na U-2012 ensaio; H2O2 adicionado para descoloramento e H2O2 presente como contaminante em Triton X-100 (De 0,22%, informações sobre o Produto: Triton X-100, www.sigmaaldrich.com). O peróxido de hidrogênio é comumente degradada pela enzima catalase. No entanto, o elevado pH do ensaio de proteínas inactivaria as catalases conhecidas. Além disso, a adição de catalase levaria à adição de proteína extra. Escolhemos a destruição química de H2O2 usando piruvato (28). A química da equação de interação piruvato-H2O2 é bem estabelecida (28,29). O piruvato destrói H2O2 à temperatura ambiente de acordo com a seguinte reação:

Residual de H2O2 na bolinha, a suspensão foi destruída pelo tratamento com 0,9 M, o piruvato (1,5 x concentração de H2O2) por 0,5 h à temperatura ambiente. Para neutralizar o contaminante H2O2 em Triton X-100, adicionou-se também piruvato extra no doseamento de proteínas (Figura 1). A adição de piruvato originou uma menor absorvância para um branco não proteico . Sugerimos que o contaminante de peróxido em Triton X-100 reage com o acetonitrilo na solução-2, dando absorvência ligeiramente maior.
a interferência da cor associada às amostras biológicas coloridas não pode ser simplesmente tomada em consideração executando um ensaio de proteína na ausência do reagente de Folina. Os rácios calculados (Abs1-Abs2) / (Abs3-Abs4) indicavam que a interferência da cor da amostra era a mais elevada para o vinho tinto (=40) e menos para a mirtilo (=6) e o beterraba (=2). Esta interferência traduziu-se em estimativas anormalmente elevadas dos níveis reais de proteínas; por exemplo, a concentração de proteínas utilizando homogeneizados de beterrabas não transformadas e transformadas (20.21 versus 2.89 mg de proteína / G de tecido, respectivamente). Além da interferência de cores, Vinho tinto e homogeneizados de beterraba e mirtilo são susceptíveis de conter substâncias que irão reagir com o reagente de Folin no ensaio U-2012 (E.G. peptídeos pequenos e açúcares complexos). Estas foram removidas por precipitação seletiva de proteínas com PCA gelada a uma concentração final de 5% (Figura 1).As curvas padrão e os seus parâmetros para as BSA não processadas e não processadas são apresentados na Figura 2. Os parâmetros derivados (A0, AM e C50) também estão listados na Tabela 1 para BSA e outras proteínas.
os resultados mostram que o erro padrão residual no modelo é baixo (0.012 a 0.048), indicando a melhor adequação dos dados à tendência da hipérbole retangular. Para comparar a informação entre várias proteínas e o seu processamento, os parâmetros foram convertidos para a concentração de absorvância = 1,0 a 650 nm (coluna direita do Quadro 1).
estes resultados mostram que a perda de proteínas (em comparação com proteínas não transformadas) nas amostras transformadas foi inferior à das amostras transformadas invertidas. Esta perda foi mais evidente no caso da tripsina e pode ser explicada com base na sua actividade auto-catalítica durante o processamento reverso. Recomendamos que o protocolo “processado” (material suplementar) seja seguido apenas para amostras biológicas que possam conter enzimas proteolíticas.
no ensaio original de Lowry (6) e na sua versão modificada U-1988 (15), apenas foi utilizada para a determinação quantitativa de proteínas a parte linear da curva-padrão obtida por representação da absorvância em função da quantidade de proteínas. No ensaio U-2012 utilizamos os dados de forma mais eficaz, ajustando uma equação de hipérbole retangular, como descrito na secção de materiais e métodos em linha com Coakley e James (20).As concentrações de proteínas em amostras desconhecidas foram calculadas por equação e em função do padrão BSA processado e da média de todas as proteínas processadas listadas no quadro 1. Este último será mais próximo de uma verdadeira estimativa para amostras biológicas que contêm uma mistura de proteínas. Estimamos as quantidades de proteína em mirtilo e beterraba em relação ao vinho tinto em aproximadamente 60 e 230 vezes, respectivamente (Tabela 2).
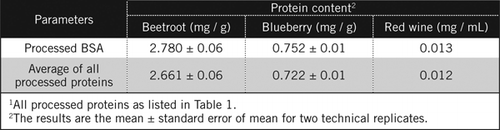
Como BSA, vinho tinto e 50% homogenates de beterraba e de mirtilo foram processados pela PCA precipitação e descoloramento por H2O2 (Figura 1). Nesta fase, as amostras biológicas foram concentradas 40 vezes para vinho tinto e 4 vezes para beterraba e mirtilo. Da mesma forma, a ASC (2 mg/mL) também se concentrou 4 vezes a 8 mg/mL. A absorvância das amostras coloridas que estava próxima da absorvância de C50 (para as BSA não transformadas) foi utilizada para calcular o teor de proteínas, tal como descrito pela equação e .
em conclusão, o ensaio de U-2012 utilizou reagentes estáveis, forneceu maior sensibilidade (mesmo para amostras biológicas coloridas) e superou a interferência induzida por cores para amostras biológicas coloridas. O ensaio de U-2012 não está limitado à parte linear da resposta entre a concentração de proteínas e a absorvância e faz um uso mais eficiente dos dados na região não linear através de um modelo de curva hiperbólica rectangular adaptado às normas utilizando procedimentos simples no Microsoft Excel.
agradecimentos
autores reconhecem a Fundação para a Investigação, Ciência e Tecnologia Nova Zelândia para o apoio financeiro (C06X0809).
interesses concorrentes
os autores não declaram interesses concorrentes.
dados Suplementares
Para visualizar os dados suplementares que acompanham este documento, por favor visite o website da revista em: www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R. S. E P. L. Pedersen. 1985. Determinação das quantidades de proteínas de Micrograma na presença de níveis de miligramas de lípidos com amido preto 10B. Anal. Bioquímica. 150:97–104.Crossref, Medline, CAS, Google Scholar
- 2. Gornall, A. G., C. J. Bardawill, and M. M. David. 1949. Determinação das proteínas séricas através da reacção do biureto. J. Biol. Chem. 177:751–766.Medline, CAS, Google Scholar
- 3. Smith, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, et al.. 1985. Medição da proteína utilizando ácido bicinchonínico. Anal. Bioquímica. 150:76–85.Crossref, Medline, CAS, Google Scholar
- 4. Bradford, M. M. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Bioquímica. 72:248–254.Crossref, Medline, CAS, Google Scholar
- 5. Zor, T. e Z. Selinger. 1996. Linearização do teste de proteína de Bradford aumenta a sua sensibilidade: estudos teóricos e experimentais. Anal. Bioquímica. 236:302–308.Crossref, Medline, CAS, Google Scholar
- 6. Lowry, O. H., N. J. Rosbrough, A. L. Farr, R. J. Randall. 1951. Medição de proteínas com o reagente de Folin fenol. J. Biol. Chem. 193:265–275.Medline, CAS, Google Scholar
- 7. Peterson, G. L. 1979. Review of the folin fenol protein quantification method of Lowry, Rosebrough, Farr and Randall. Anal. Bioquímica. 100:201–220.Crossref, Medline, CAS, Google Scholar
- 8. Sapan, C. V., R. L. Lundablad, and N. C. Price. 1999. Técnicas de ensaio de proteínas colorimétricas. Biotechnol. Appl. Bioquímica. 29:99–108.Medline, CAS, Google Scholar
- 9. Okutucu, B., A. Dınçer, Ö. Habib, and F. Zıhnıoglu. 2007. Comparação de cinco métodos para determinação da concentração total de proteínas plasmáticas. J. Biochem. Biophys. Methods 70: 709-711.Crossref, Medline, CAS, Google Scholar
- 10. Kresge, N., R. D. Simoni, and R. L. Hill. 2005. The most highly cited paper in publishing history: protein determination by Oliver H. Lowry. J. Biol. Chem. 25:280.Google Scholar
- 11. Everette, J. D., Q. M. Bryant, A. M. Green, Y. A. Abbey, G. W. Wangila, and R. B. Walker. 2010. Estudo completo da reatividade de várias classes de compostos para o reagente Folin-Ciocalteu. J. Agric. Comida Química. 58:8139–8144.Crossref, Medline, CAS, Google Scholar
- 12. Eichberg, J. and L. C. Mokrasch. 1969. Interferência por lípidos oxidados na determinação da proteína pelo procedimento de Lowry. Anal. Bioquímica. 30:386–390.Crossref, Medline, CAS, Google Scholar
- 13. Dulley, Jr E P. A. Grieve. 1975. Uma técnica simples para eliminar a interferência dos detergentes no método de Lowry de determinação de proteínas. Anal. Bioquímica. 64:136–141.Crossref, Medline, CAS, Google Scholar
- 14. Brillouet, J.-M., M.-P. Belleville, and M. Moutounet. 1991. Possíveis complexos proteico-polissacáridos em vinhos tintos. Manha. J. Enol. Tritic. 42:150–152.CAS, Google Scholar
- 15. Upreti, G. C., R. A. Ratcliff, and P. C. Riches. 1988. Estimativa de proteínas nos tecidos que contêm níveis elevados de lípidos: modificações do método de Lowry de determinação de proteínas. Anal. Bioquímica. 168:421–427.Crossref, Medline, CAS, Google Scholar
- 16. Upreti, G. C., C. Davis, and J. Oliver. 1991. Preparação de homogeneizados representativos dos tecidos biológicos: efeito do sal na extracção de proteínas. Anal. Bioquímica. 198:298–301.Crossref, Medline, CAS, Google Scholar
- 17. Smith, M. R., M. H. Penner, S. E. Bennett, and A. T. Bakalinsky. 2011. Ensaio colorimétrico quantitativo para a proteína Total aplicada ao Pinot Noir De Vinho Tinto. J. Agric. Comida Química. 59:6871–6876.Crossref, Medline, CAS, Google Scholar
- 18. Wigand, P., S. Tenzer, H. Schild, and H. Decker. 2009. Análise da composição proteica do Vinho Tinto em comparação com os vinhos rosados e brancos por electroforese e cromatografia líquida de alta pressão-espectrometria de massa (HPLC-MS). J. Agric. Comida Química. 57:4328–4333.Crossref, Medline, CAS, Google Scholar
- 19. Upreti, G. C., Y. Wang, A. Sharrock, N. Feisst, M. Davy, and B. Jordan. 2009. A stable and sensitive protein assay (u-2009 modified assay) for colored biological samples. ComBiol., Programa Final Da Nova Zelândia, Dezembro De 2009. University of Canterbury, Christchurch, New Zealand.Google Scholar
- 20. Coakley, W. T. and C. J. James. 1978. Uma simples transformação linear para a curva de calibração da proteína Folin-Lowry até 1, 0 mg/mL. Anal. Bioquímica. 85:90–97.Crossref, Medline, CAS, Google Scholar
- 21. Pinheiro, J. C. and D. M. Bates. 2000. Mixed-Effects Models in s and S-PLUS, Statistics and Computing Series. Springer-Verlag, New York, NY.Google Scholar
- 22. R Equipa De Desenvolvimento Central. 2009. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, ISBN 3-900051-07-0, URL http://www.R-project.org.Google Scholar
- 23. Khachik, F., G. R. Beecher, J. T. Vanderslice, and G. Sulco. 1988. Artefactos cromatográficos líquidos e distorção de pico: interacção amostra-solvente na separação de carotenoides. Anal. Chem. 60:807–811.Crossref, Medline, CAS, Google Scholar
- 24. Cernik, A. A. 1970. Determinação do chumbo quelatado com ácido etilenodiaminetetra-acético no sangue após precipitação de proteínas com ácido perclórico. Britanico. J. Industry Med. 27:40–42.Medline, CAS, Google Scholar
- 25. Moughan, P. J., A. J. Darragh, W. C. Smith, C. A. Pontas. 1990. Ácidos perclóricos e tricloroacéticos como precipitantes de proteínas na digesta ileal endógena do rato. J. Sci. Agric. 52:13–21.Crossref, CAS, Google Scholar
- 26. Galbács, Z. M. and L. J. Csányi. 1983. Decomposição alcalina de peróxido de hidrogénio. J. Chem. Soc., Dalton Trans. 11:2353–2357.Crossref, Google Scholar
- 27. Scopes, R. K. 1988. Purificação de proteínas: Principles and Practice, Second Ed. Springer-Verlag New York Inc., New York, NY.Google Scholar
- 28. Upreti, G. C., K. Jensen, R. Munday, D. M. Duganzich, R. Aurelina, e J. F. Smith. 1998. Estudos sobre a actividade da aminoácido aromático oxidase em espermatozoa ram: papel do piruvato como antioxidante. Anim. Reprovação. Ciência. 51:275–287.Crossref, Medline, CAS, Google Scholar
- 29. Holleman, M. A. F. 1904. Notice sur l’action de l’EAU oxigenee sur les acids α-cetoniques et sur les dicetones 1.2. Recl. Trav. Chim. Pays-Bas Belg. 23:169–172.Crossref, CAS, Google Scholar