kvantitative målinger af fysiologiske træk såsom f.eks. Selvom der er udviklet adskillige assays til måling af proteinindhold, herunder de kolorimetriske assays af Amido Black (1), Biuret (2), Bicinchoninsyre (3) og Coomassie Blue (4,5), er Lavtassayet (6) eller dets modifikationer (7,8) mere almindeligt anvendt end andre assays (9). Analysen er enkel, følsom og præcis og er den mest citerede (10) procedure til kvantitativ proteinbestemmelse.
en lang række forbindelser, der reagerer med Folin-Ciocalteu phenol (Folin ‘ s) reagens (11), er en kilde til potentiel interferens i analyser af lavt og modificeret lavt proteinindhold. Heldigvis er korrektioner gennem et passende emne tilstrækkeligt for de fleste forbindelser (6,7) undtagen lipider (12), vaskemidler (13) og farvede stoffer (14). Vanskeligheder ved analyse af proteiner i nærvær af lipider og vaskemidler (anvendt til opløseliggørelse af fedtvæv, myelin og skeletmuskler) blev overvundet af det modificerede Lavry assay (15; omtalt i dette papir som u-1988 assay, 16). Farveinterferens ved bestemmelse af proteinindholdet i rødvin (14,17,18) blev overvundet ved anvendelse af omfattende kromatografi. Ovenstående fremgangsmåde er besværlig og ikke særlig praktisk til håndtering af et stort antal prøver. Ingen af de kendte proteinanalyser var egnede til at måle proteiner i farvede biologiske prøver, f. eks., farvede frugter og grøntsager, rødvin, pigmenterede mikrober og drøvtyggere galde.
vores udvikling af U-2012-analysen fra sine forgængere U-1988 og Lavtassayet har opnået tre store fordele (i) bekvemmelighed gennem stabilitet af reagensformuleringerne, (ii) måling af protein i både farveløse og farvede biologiske prøver uden at gå på kompromis med følsomheden og (iii) analyse af proteiner i meget lave koncentrationer. Denne nye analyse vil være anvendelig til kvantitativ bestemmelse af protein i både farveløse og farvede biologiske prøvehomogenater, inklusive dem, der er rige på lipider (f.eks. avocado) og dem, der er vanskelige at homogenisere.
- materialer og metoder
- biologiske prøver – rødbeder, blåbær og rødvin
- kemiske reagenser
- forbedringer af U-1988-assay
- u-2012-analysen
- estimering af farveinterferenser i U-2012-analysen
- standardkurven og dens parametre
- beregning af proteinindholdet i homogenaterne
- resultater og diskussion
- forbedringer i U-1988-analysen
- protein estimering i farvede biologiske prøver
- proteinekstraktion
- Eliminer interfererende stoffer
- Standardkurver og deres parametre
- proteinindhold i farvet homogenat
- anerkendelser
- konkurrerende interesser
- supplerende data
materialer og metoder
biologiske prøver – rødbeder, blåbær og rødvin
rødbeder og blåbærhomogenater blev fremstillet som beskrevet i det supplerende materiale. Rødvin krævede ikke proteinekstraktion før u-2012-analysen.
kemiske reagenser
alle kemiske reagenser undtagen natriumhypochlorit og perchlorsyre (PCA) blev opnået fra Sigma eller Sigma-Aldrich (St Louis, MO. USA). Natriumhypochlorit var fra Acros Organic, ny Jersey, USA. PCA blev opnået fra BDH (England).
forbedringer af U-1988-assay
skift fra carbonat til phosphatbuffer ved pH 12,0 forbedret reagensstabilitet og gav en lille stigning i følsomhed. Acetonitril blev introduceret for at undgå vaskemiddelinducerede bobler. NaOH erstattede KOH for at undgå udfældning i proteinanalysen. Derudover blev effektiviteten forbedret ved at kombinere forskellige komponenter i Lavreagensen i en reagensblanding.
u-2012-analysen
alle detaljer om u-2012-analysen findes i supplerende materiale. Protokollen, kort opsummeret i Figur 1, beskriver forarbejdningen af rødvin og homogenaterne af rødbeder og blåbær og inkluderer forbedringerne af U-1988-analysen. U-2012-analysen blev anvendt til uforarbejdede, forarbejdede og omvendte forarbejdede (H2O2-behandling efterfulgt af TCA-eller PCA-udfældning) proteiner. Analyser blev udført på BSA, kulsyreanhydrase, cytokrom C, isocitrat dehydrogenase, lysosyme og trypsin til udvikling af standardkurver og i farvede biologiske prøver. Bestemmelsen af proteiner i de biologiske prøver blev udført ved kalibrering til passende standardkurver.
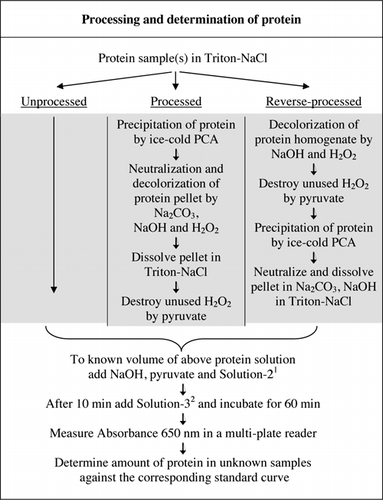
1opløsning-2 indeholdt kobbersulfat (CuSO4.5h2o), Na-K-tartrat, SDS og acetonitril i 100 mM phosphatbuffer (pH 12,0). 2For opløsning-3 blev Folin-Ciocalteus phenolreagens fortyndet 1:1 med deioniseret vand lige før brug.
estimering af farveinterferenser i U-2012-analysen
farveinterferens blev bestemt ved at sammenligne absorptionsevnen fra forarbejdede og uforarbejdede rødbeder -, blåbær-og rødvinsprøver både med og uden brug af Folins reagens som beskrevet i Figur 1. Forholdet blev brugt til at bestemme omfanget af interferens, hvor Abs1 er absorbans af uforarbejdede prøver med Folins reagens; Abs2 er absorbans af uforarbejdede prøver uden Folins reagens; Abs3 er absorbans af forarbejdede prøver med Folins reagens; og Abs4 er absorbans af forarbejdede prøver uden Folins reagens.
standardkurven og dens parametre
opløsning-1b og 1C beskrevet i Opskriftsafsnittet i det vedhæftede supplerende materiale blev brugt til udvikling af standardkurverne. Koncentrationen af BSA og de tilsvarende absorbansværdier blev afbildet ved hjælp af en røntgen-scatter-graf. Formen af denne graf (figur 2) viser et mættende respons ved højere koncentrationer med et meget begrænset indledende lineært respons. Dette var en foretrukken kurveform rapporteret tidligere (20). Oprindeligt blev dette modelleret ved hjælp af en eksponentiel form (19), men senere undersøgelser viste, at en rektangulær hyperbola gav en forbedret tilpasning til responsen, især ved lavere koncentrationer. Denne sidstnævnte form er nu standardiseret, og følgende tre-parameterligning blev brugt til at beskrive absorbans-proteinkoncentrationsforholdet:

Conc = proteinkoncentration,
A = absorbans ved Conc,
A0 = absorbans ved nulkoncentration,
AM= absorbans ved maksimal koncentration,
C50= koncentration, der giver absorbans
(AM + A0)/2.
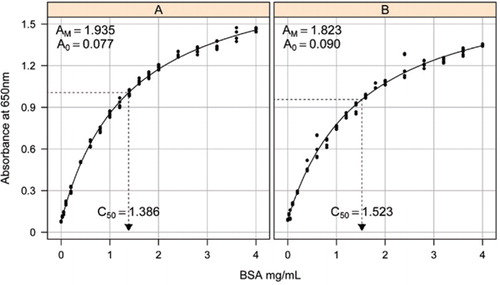
estimering af standardkurven mellem absorbansen (650 nm) og koncentrationerne af BSA-protein blev foretaget på tre tekniske replikater til (A) det uforarbejdede proteinassay (parametre A0= 0,077, Am=1,935, C50=1.386), og B) den forarbejdede proteinanalyse (parametre A0=0,90, Am=1,823, C50=1,523). En rektangulær hyperbolamodel (ligning ) blev monteret på de observerede data. Parameterestimater identificeres i kurverne med C50-estimeringen på h-aksen (prikket sort linje).
Parameter A0 blev eksperimentelt bestemt, mens AM og C50 blev estimeret ved hjælp af Microsoft værktøjskasse add-in Solver funktion. Et forsøgssæt med parametre blev brugt til at beregne den modellerede absorbans ved hver af standardkoncentrationerne (Conc) ved hjælp af ligning . Solver blev derefter befalet at minimere den resterende standardafvigelse mellem den målte og modellerede absorbans for standardindstillingen ved at justere AM og C50.
vi observerede forholdet mellem absorbans og koncentration for at udvise en ikke-lineær kurve over hele koncentrationsområdet, hvilket sandsynligvis skyldes en komponent af lysspredning, der stiger, når koncentrationen af protein stiger ved måling af absorbans. En dårlig lineær pasform ved lav absorbans blev også rapporteret af Coakley og James (20).
beregning af proteinindholdet i homogenaterne
analyser blev udført på forarbejdet og uforarbejdet rødvin og homogenater af rødbeder og blåbær. BSA og andre proteinprøver blev behandlet identisk for passende standardkurver til bestemmelse af A0 -, AM-og C50-værdier. Disse parametre blev derefter brugt til at konvertere prøveabsorbans (A) til proteinkoncentration i hvert homogenat ved anvendelse af:

fordi ligningen har en mættende form, reduceres følsomheden som absorbans (A) og dermed koncentration øges. Fejl i proteinestimeringer kan minimeres ved at justere koncentrationerne af homogenater i analysen, så de ikke overstiger C50-værdien for meget.
Homogenatkonc-værdien blev derefter omdannet til vævsproteinkoncentration (Vævskonc i mg/g væv) ved hjælp af følgende formel:

hvor homogenat Conc (i mg protein/mL) er blevet korrigeret for enhver prækoncentration eller fortynding under analysen. Homogenatprocent var 100 g væv homogeniseret til et samlet volumen på 200 mL (i vores tilfælde 50%).
i en separat undersøgelse blev der monteret en rektangulær hyperbolamodel ved anvendelse af pakken non linear blandede effekter (NLME) (21) I R (22) (figur 2). Hver BSA-opløsning, fremstillet uafhængigt i laboratoriet, blev modelleret som en tilfældig effekt med en fælles A0, men forskellige AM-og C50-koefficienter. Dette modellerer hierarkiet af biologiske prøver replikater og tekniske assay replikater.
resultater og diskussion
forbedringer i U-1988-analysen
begrænsningen af U-1988 og Lavry-analysen er ustabiliteten af det carbonatbaserede reagens. Carbonatbufferen (pH 11,4 ved 2% = 188,7 mM) i U-1988 blev erstattet med 40 mM phosphat ved pH-værdier i området fra 11,4 til 12,5. Indledende skråninger fra standardkurverne for proteinanalysen under anvendelse af BSA ved 0,5 mg BSA/ mL og 1,0 mg BSA / mL blev beregnet. De oprindelige skråninger med fosfatbuffere ved pH 11,4 og ved dens optimale pH 12,0 var 99 H10-6 og 197 H10-6 henholdsvis. Hældningen for carbonatbufferen (pH 11,4) var 162 H10-6.Da hældningsværdien er en direkte indikation af assayfølsomhed, fosfatbuffer (pH 12.0) blev valgt til at erstatte carbonatbuffer, hvilket gav en 25% stigning i følsomhed.
større stabilitet blev opnået ved at øge koncentrationen af phosphatbufferen til 100 mM. den resulterende phosphat/CuSO4/Na-K-tartratopløsning var stabil ved stuetemperatur i to uger, betydeligt længere end carbonat/CuSO4/Na-K-tartratopløsning, som skal fremstilles dagligt før proteinanalyse. Til alle fremtidige eksperimenter blev 100 mM fosfat (pH 12,0) anvendt til fremstilling af CuSO4/Na-K-tartratopløsningen. Vi mener, at udskiftning af carbonat med fosfat vil forbedre bekvemmeligheden ved U-2012-analysen.
vaskemiddelinducerede bobler bliver en vigtig fejlkilde i absorbansmålinger, når du bruger en pladelæser med flere brønde (ikke et problem med kuvetter). Disse bobler blev reduceret betydeligt ved tilsætning af et antal polære opløsningsmidler (f.eks. acetone, acetonitril, ethanol og methanol). Acetonitril, den mest polære af disse opløsningsmidler (23) blev valgt for dets effektivitet og inkluderet i opløsning-2 (Se figur 1 billedtekst og opskrifter sektion af supplerende materiale).
phosphatbuffer, CuSO4, Na-K-tartrat, SDS og acetonitril kan tilsættes individuelt, og rækkefølgen af deres tilsætning påvirker ikke den resulterende absorbans. Brug af en forblandet opløsning forbedrer imidlertid bekvemmeligheden yderligere, især når et stort antal prøver skal analyseres. Vi grupperede derfor disse komponenter i analyseblandingen i opløsning-2 (Figur 1). En sådan forblandet opløsning var ikke mulig for den oprindelige Lavry assay (6) på grund af ustabilitet af carbonatopløsningen. Forsøget på at inkludere opløsning-3 i opløsning-2 resulterede i dramatisk reduktion i udviklingen af blå farve og blev ikke overvejet yderligere.
protein estimering i farvede biologiske prøver
proteinekstraktion
proteiner fra rødbeder og blåbær blev ekstraheret i Triton 100-NaCl opløsning med mild homogenisering. Sådanne homogenater bevarer deres aktiviteter (15). Denne ekstraktion var ikke nødvendig for rødvin.
Eliminer interfererende stoffer
for farvede prøver er det nødvendigt at fjerne interferensen på grund af den iboende prøvefarve og andre ikke-proteinstoffer, der reagerer med proteinreagenserne før kolorimetrisk proteinanalyse. Nyheden af U-2012 er at udtænke en affarvningsprotokol, der er kompatibel med et kolorimetrisk proteinassay.
affarvning af farvede pigmenter med natriumhypochlorit eller H2O2 og selektiv udfældning af proteiner med PCA eller TCA blev overvejet til fjernelse af interfererende stoffer. Natriumhypochlorit, H2O2, TCA og PCA blev evalueret for deres kompatibilitet med U-2012-analysen ved anvendelse af BSA som testprotein. Mellem natriumhypochlorit og H2O2 var kun H2O2 kompatibel, da der blev dannet et bundfald i nærvær af hypochlorit. Proteiner udfældet af TCA eller PCA kan analyseres af U-2012 efter tilstrækkelig neutralisering af restsyre i pelleten. PCA ‘ s overlegenhed over TCA for proteinudfældning er rapporteret (24,25). I modsætning hertil afslørede i vores sammenlignende evaluering lignende C50-værdier for PCA (1.395) og TCA (1.400). Vi foretrak PCA, fordi den er let tilgængelig som en færdiglavet opløsning (70% v/v) og derfor let fortyndet til den krævede styrke. TCA er et hygroskopisk fast stof, der er vanskeligt at veje præcist på grund af dets variable vandindhold.
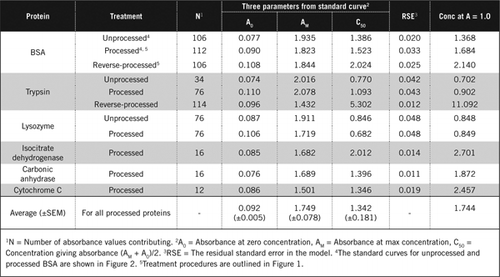
der er to mulige måder at kombinere PCA og H2O2 på. For” forarbejdede ” proteiner blev PCA-behandling efterfulgt af H2O2-behandling og for “reverse-processed” protein, H2O2-behandling forud for PCA-udfældning. Fordelene ved at anvende’ forarbejdet ‘ protein var fjernelsen af et antal interfererende stoffer i supernatanten og den mulige inaktivering af proteolytiske stoffer under prøveforberedelse. Dette blev bekræftet ved analyse af behandlet og omvendt behandlet trypsin og BSA (Se tabel 1). Kun forarbejdede prøver blev brugt til at bestemme det faktiske proteinindhold i farvede biologiske prøver
både PCA-og H2O2-behandlinger af farvede prøver var nødvendige for interferenseliminering i U-2012-analyse. Syreudfældning alene af de farvede prøver fjernede ikke interferens fuldstændigt. Med alle de farvede prøver blev noget farve kasseret i supernatanten, men pellets blev også farvet. Farve blev elimineret fra pellets ved H2O2-behandling. Alkaliske betingelser var nødvendige for både effektiv affarvning ved H2O2 (26) og farveudviklingen ved Folins reagens for at sikre, at proteinniveauer måles korrekt. Selvom både NaOH og KOH kunne give den krævede alkalinitet, var kun NaOH kompatibel med U-2012-analysen. Et bundfald blev dannet i nærvær af KOH. I pellets blev PCA neutraliseret ved hjælp af Na2CO3 og NaOH (27). Yderligere NaOH blev tilføjet under analysen; det optimerede volumen var mellem 50 og 70 liter (60 liter blev rutinemæssigt brugt); se figur 1.
rødbeder, blåbær og rødvin blev affarvet med 15 liter på 30% H2O2, der tog henholdsvis 0,5 og 2 timer ved 50 liter C og stuetemperatur. Tyve mikroliter på 30% H2O2 til 1 h ved 50 liter C blev brugt til at klare stærkere farvede prøver. Iltning af stoffer som sukker, der er bundet til proteiner af H2O2 ved 50 liter C, synes kritisk, da behandling ved stuetemperatur overvurderer proteinindholdet. I tilfælde af rødbeder reducerede 50 kg c-behandling det tilsyneladende proteinestimat til 14% af det uforarbejdede, mens behandling ved stuetemperatur kun halverede dette skøn.
det fremgik af de kolorimetriske analyser, der blev udført efter brintoverilte-behandling, at noget H2O2 ikke blev anvendt i affarvningen. I en sådan prøve blev Endefarven på Lavry-analysen delvist ødelagt. Det var derfor nødvendigt at ødelægge det resterende brintoverilte før proteinanalyse. Der er to kilder til H2O2 i U-2012-analysen; H2O2 tilføjet til affarvning og H2O2 til stede som et forurenende stof i Triton 100 (0,22%, produktinformation: Triton 100, www.sigmaaldrich.com brintoverilte nedbrydes almindeligvis af katalasen. Imidlertid ville proteinassayets høje pH inaktivere kendte katalaser. Tilsætning af katalase ville også føre til tilsætning af ekstra protein. Vi valgte kemisk destruktion af H2O2 ved hjælp af pyruvat (28). Kemien i pyruvat-H2O2 interaktionsligningen er veletableret (28,29). Pyruvat ødelægger H2O2 ved stuetemperatur ifølge følgende reaktion:

resterende H2O2 i pillesuspensionen blev ødelagt ved behandling med 0,9 m pyruvat (1,5 gange koncentration af H2O2) i 0,5 timer ved stuetemperatur. For at modvirke den kontaminerende H2O2 i Triton H-100 blev der også tilsat ekstra pyruvat i proteinanalysen (Figur 1). Tilsætningen af pyruvat gav lavere absorbans for et ikke-proteinemne . Vi foreslår, at forureningen i Triton H-100 reagerer med acetonitril i opløsning-2, hvilket giver en lidt højere absorptionsevne.
farveinterferens forbundet med de farvede biologiske prøver kan ikke blot tages i betragtning ved at køre et proteinassay i fravær af Folins reagens. De beregnede forhold (Abs1-Abs2) / (Abs3-Abs4) indikerede, at interferens fra prøvefarve var den højeste for rødvin (=40) og mindre for blåbær (=6) og rødbeder (=2). Denne interferens blev oversat til de unormalt høje estimater af ægte proteinniveauer; for eksempel koncentrationen af protein ved anvendelse af uforarbejdede og forarbejdede rødbederhomogenater (henholdsvis 20,21 versus 2,89 mg protein / g væv). Ud over farveinterferens vil rødvin og homogenater af rødbeder og blåbær sandsynligvis indeholde stoffer, der vil reagere med Folins reagens i U-2012-analysen (e.g., små peptider og komplekse sukkerarter). Disse blev fjernet ved selektivt udfældning af proteiner med iskold PCA i en endelig koncentration på 5% (Figur 1).
Standardkurver og deres parametre
Standardkurver for uforarbejdet og forarbejdet BSA er vist i figur 2. De afledte parametre (A0, AM og C50) er også anført i tabel 1 for BSA og andre proteiner.
resultaterne viser, at den resterende standardfejl i modellen er lav (0,012 til 0,048), hvilket indikerer, at dataene passer bedre til den rektangulære hyperbolatrend. Til sammenligning af information mellem forskellige proteiner og deres behandling blev parametrene konverteret til koncentrationen for absorbans = 1,0 ved 650 nm (højre kolonne i tabel 1).
disse resultater viser, at tab af protein (sammenlignet med uforarbejdet protein) i forarbejdede prøver var mindre end de omvendt forarbejdede prøver. Dette tab var mere tydeligt i tilfælde af trypsin, og det kan forklares på grundlag af dets auto-katalytiske aktivitet under omvendt behandling. Vi anbefaler, at den ‘forarbejdede’ protokol (supplerende materiale) kun følges for biologiske prøver, der sandsynligvis indeholder proteolytiske stoffer.
i den oprindelige Lavry assay (6) og dens modificerede version U-1988 (15) blev kun den lineære del af standardkurven opnået ved at plotte absorbansen mod mængden af protein anvendt til kvantitativ bestemmelse af protein. I U-2012-analysen bruger vi dataene mere effektivt ved at montere en rektangulær hyperbolaligning som beskrevet i afsnittet Materialer og metoder i tråd med Coakley og James (20).
proteinindhold i farvet homogenat
proteinkoncentrationer i ukendte prøver blev beregnet ved ligning og mod forarbejdet BSA-standard og gennemsnittet af alle forarbejdede proteiner anført i tabel 1. Sidstnævnte vil være tættere på et sandt skøn for biologiske prøver, der indeholder en blanding af proteiner. Vi anslog mængderne af protein i blåbær og rødbeder i forhold til rødvin til henholdsvis 60 og 230 gange (tabel 2).
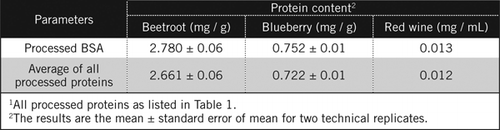
ligesom BSA blev rødvin og 50% homogenater af rødbeder og blåbær forarbejdet ved PCA-udfældning og affarvning med H2O2 (Figur 1). På dette stadium blev de biologiske prøver koncentreret 40 gange for rødvin og 4 gange for rødbeder og blåbær. Tilsvarende blev BSA (2 mg/mL) også koncentreret 4 gange til 8 mg/mL. Absorbansen af de farvede prøver, der var tæt på absorbansen for C50 (for uforarbejdet BSA) blev brugt til at beregne proteinindholdet, som beskrevet ved ligning og .
afslutningsvis har U-2012-analysen anvendt stabile reagenser, forudsat forbedret følsomhed (selv for farveløse biologiske prøver) og overvinde farveinduceret interferens for farvede biologiske prøver. U-2012-analysen er ikke begrænset til den lineære del af responsen mellem proteinkoncentration og absorbans og gør mere effektiv brug af data i det ikke-lineære område gennem en rektangulær hyperbolsk kurvemodel tilpasset standarderne ved hjælp af enkle procedurer inden for Microsoft.
anerkendelser
forfattere anerkender Fonden for forskning, videnskab og teknologi for økonomisk støtte (C06H0809).
konkurrerende interesser
forfatterne erklærer ingen konkurrerende interesser.
supplerende data
for at se de supplerende data, der ledsager dette papir, kan du besøge tidsskriftets hjemmeside på: www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R. S. og P. L. Pedersen. 1985. Bestemmelse af mikrogram mængder protein i nærvær af milligram niveauer af lipid med amido black 10B. Anal. Biochem. 150:97–104.Crossref, Medline, CAS, Google Scholar
- 2. Gornall, A. G., C. J. Bardavill og M. M. David. 1949. Bestemmelse af serumproteiner ved hjælp af biuret-reaktionen. J. Biol. Chem. 177:751–766.Medline, CAS, Google Scholar
- 3. Det er en af de mest almindelige årsager til, at der er tale om en alvorlig sygdom, der kan føre til, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en alvorlig risiko for, at der opstår en risiko for, at der opstår en alvorlig risiko for, at der opstår problemer.. 1985. Måling af protein ved anvendelse af bicinchoninsyre. Anal. Biochem. 150:76–85.Crossref, Medline, CAS, Google Scholar
- 4. Bradford, M. M. 1976. En hurtig og følsom metode til kvantificering af mikrogram mængder protein under anvendelse af princippet om protein-farvestof binding. Anal. Biochem. 72:248–254.Crossref, Medline, CAS, Google Scholar
- 5. T. og Selinger. 1996. Linearisering af Bradford-proteinanalysen øger dens følsomhed: teoretiske og eksperimentelle undersøgelser. Anal. Biochem. 236:302–308.Crossref, Medline, CAS, Google Scholar
- 6. R. J. Rosbrough, A. L. Farr og R. J. Randall. 1951. Proteinmåling med Folinphenolreagensen. J. Biol. Chem. 193:265–275.Medline, CAS, Google Scholar
- 7. Peterson, G. L. 1979. Gennemgang af folin phenolproteinkvantificeringsmetoden for Lavry, Rosebrough, Farr og Randall. Anal. Biochem. 100:201–220.Crossref, Medline, CAS, Google Scholar
- 8. Sapan, C. V., R. L. Lundablad og N. C. Price. 1999. Kolorimetriske protein assay teknikker. Biotechnol. Appl. Biochem. 29:99–108.Medline, CAS, Google Scholar
- 9. Okutucu, B., A. D. Habib og F. kr. 2007. Sammenligning af fem metoder til bestemmelse af total plasmaproteinkoncentration. J. Biochem. Biofys. Metoder 70: 709-711.Crossref, Medline, CAS, Google Scholar
- 10. Kresge, N., R. D. Simoni og R. L. Hill. 2005. Det mest citerede papir i forlagshistorien: proteinbestemmelse af Oliver H. Lavry. J. Biol. Chem. 25:280.Google Scholar
- 11. Det er en af de mest populære og mest populære destinationer i verden. 2010. Grundig undersøgelse af reaktivitet af forskellige sammensatte klasser mod Folin-Ciocalteu-reagenset. J. Agric. Mad Chem. 58:8139–8144.Crossref, Medline, CAS, Google Scholar
- 12. Eichberg, J. Og L. C. Mokrasch. 1969. Interferens af iltede lipider i bestemmelsen af protein ved Lavtproceduren. Anal. Biochem. 30:386–390.Crossref, Medline, CAS, Google Scholar
- 13. Dulley, J. R. og P. A. sørger. 1975. En simpel teknik til eliminering af interferens fra vaskemidler i den lave metode til proteinbestemmelse. Anal. Biochem. 64:136–141.Crossref, Medline, CAS, Google Scholar
- 14. Brillouet, J.-M., M.-P. Belleville og M. Moutounet. 1991. Mulige protein-polysaccharidkomplekser i røde vine. Er. J. Enol. Vitic. 42:150–152.CAS, Google Scholar
- 15. Upreti, G. C., R. A. Ratcliff og P. C. Riches. 1988. Proteinestimering i væv, der indeholder høje niveauer af lipid: modifikationer til Lavry metode til proteinbestemmelse. Anal. Biochem. 168:421–427.Crossref, Medline, CAS, Google Scholar
- 16. Upreti, G. C., C. Davis og J. Oliver. 1991. Fremstilling af repræsentative homogenater af biologiske væv: virkning af salt på proteinekstraktion. Anal. Biochem. 198:298–301.Crossref, Medline, CAS, Google Scholar
- 17. Smith, M. R., M. H. Penner, S. E. Bennett og A. T. Bakalinsky. 2011. Kvantitativ kolorimetrisk analyse for Total Protein anvendt på rødvin Pinot Noir. J. Agric. Mad Chem. 59:6871–6876.Crossref, Medline, CAS, Google Scholar
- 18. H. Schild og H. Decker. 2009. Analyse af proteinsammensætning af rødvin i sammenligning med ros og hvide vine ved elektroforese og højtryksvæskekromatografi-massespektrometri (HPLC-MS). J. Agric. Mad Chem. 57:4328–4333.Crossref, Medline, CAS, Google Scholar
- 19. Det er en af de mest populære ting i verden. 2009. Et stabilt og følsomt proteinassay (u-2009 modificeret assay) til farvede biologiske prøver. ComBiol. Danmarks Endelige Program December 2009. University of Canterbury, Christchurch, Danmark.Google Scholar
- 20. Coakley, C. J. James og C. J. James. 1978. En simpel lineær transformation for Folin-Lavryproteinkalibreringskurven til 1,0 mg/mL. Anal. Biochem. 85:90–97.Crossref, Medline, CAS, Google Scholar
- 21. Pinheiro, J. C. og D. M. Bates. 2000. Blandede effekter modeller i S og S-PLUS, statistik og Computing serien. Springer-Verlag, Ny, Ny.Google Scholar
- 22. R Udvikling Core Team. 2009. R: et sprog og miljø for statistisk databehandling. R Foundation for Statistical Computing, Vienna, Østrig, ISBN 3-900051-07-0, URL http://www.R-project.org.Google Scholar
- 23. Khachik, F., G. R. Beecher, J. T. Vanderslice og G. Fure. 1988. Væskekromatografiske artefakter og spidsforvrængning: prøve-opløsningsmiddelinteraktion i adskillelsen af carotenoider. Anal. Chem. 60:807–811.Crossref, Medline, CAS, Google Scholar
- 24. Cernik, AA 1970. Bestemmelse af blychelateret med ethylendiaminetetra-eddikesyre i blod efter udfældning af protein med perchlorsyre. Brite. J. Industri Med. 27:40–42.Medline, CAS, Google Scholar
- 25. Moughan, P. J., A. J. Darragh, C. A. Smith og C. A. Butts. 1990. Perchlorsyre og trichloreddikesyre som præcipitanter af protein i endogen ileal digesta fra Rotten. J. Sci. Mad Agric. 52:13–21.Crossref, CAS, Google Scholar
- 26. Galb og L. J. C. 1983. Alkali-induceret nedbrydning af brintoverilte. J. Chem. Soc. Dalton Trans. 11:2353–2357.Crossref, Google Scholar
- 27. Scopes, R. K. 1988. Proteinrensning: principper og praksis, anden udgave. Springer-Verlag Danmark Inc., Ny, Ny.Google Scholar
- 28. Det er et af de mest kendte og mest kendte steder i verden. 1998. En af de mest almindelige årsager til denne sygdom er, at en person er i stand til at bestemme, om han eller hun er i stand til at blive gravid. Anim. Reprod. Sci. 51:275–287.Crossref, Medline, CAS, Google Scholar
- 29. Holleman, M. A. F. 1904. Læg mærke til, hvordan man bruger ilt på syre og syre 1.2. Recl. Trav. Chim. Pays-Bas Belg. 23:169–172.Crossref, CAS, Google Scholar