a fiziológiai tulajdonságok, például az enzimaktivitás kvantitatív méréseit gyakran milligramm fehérjénként fejezik ki. Bár számos vizsgálatot fejlesztettek ki a fehérjetartalom mérésére, beleértve az Amido Black (1), A Biuret (2), A Bicinchoninic Acid (3) és a Coomassie Blue (4,5) kolorimetriás vizsgálatait, a Lowry-tesztet (6) vagy annak módosításait (7,8) gyakrabban használják, mint más vizsgálatokat (9). A Lowry-vizsgálat egyszerű, érzékeny és pontos, és a legtöbbet idézett (10) eljárás a mennyiségi fehérje meghatározására.
a Folin-Ciocalteu fenol (Folin) reagenssel (11) reakcióba lépő vegyületek széles választéka potenciális interferencia forrása a Lowry és a módosított Lowry protein vizsgálatokban. Szerencsére a megfelelő vakpróbával történő korrekció elegendő a legtöbb vegyülethez (6,7), kivéve a lipideket (12), a mosószereket (13) és a színezett anyagokat (14). A lipidek és detergensek jelenlétében (a zsírszövet, a mielin és a vázizmok szolubilizálásában használt) fehérjék vizsgálatának nehézségeit a módosított Lowry assay (15; ebben a tanulmányban U-1988 assay, 16). A vörösbor (14,17,18) fehérjetartalmának meghatározásakor a színinterferenciát kiterjedt kromatográfiával sikerült leküzdeni. A fenti megközelítés nehézkes és nem túl praktikus nagyszámú minta kezelésére. Az ismert fehérjetesztek egyike sem volt alkalmas színes biológiai mintákban lévő fehérjék mérésére, pl., színes gyümölcsök és zöldségek, vörösbor, pigmentált mikrobák és kérődző epe.
az U-2012 teszt kifejlesztése elődeitől, az U-1988-tól és a Lowry-vizsgálattól három fő előnyt ért el (i) a reagenskészítmények stabilitása révén a kényelem, (ii) a fehérje mérése színtelen és színes biológiai mintákban az érzékenység veszélyeztetése nélkül, és (iii) a fehérjék vizsgálata nagyon alacsony koncentrációban. Ez az új vizsgálat alkalmazható a fehérje mennyiségi meghatározására mind a színtelen, mind a színes biológiai minta homogenizátumokban, beleértve a lipidekben gazdagokat (például avokádót) és azokat, amelyeket nehéz homogenizálni.
- anyagok és módszerek
- biológiai minták – cékla, áfonya és vörösbor
- kémiai reagensek
- az U-1988 assay javításai
- az U-2012 vizsgálat
- a színinterferenciák becslését az U-2012-es vizsgálatban
- a standard görbék kidolgozásához a mellékelt kiegészítő anyag receptek részében leírt standard görbét és paramétereit,
- a homogenizátumok fehérjetartalmának kiszámítása
- eredmények és vita
- fejlesztések az U-1988-as vizsgálatban
- fehérje becslés színes biológiai mintákban
- fehérje extrakció
- interferáló anyagok eltávolítása
- Standard görbék és paramétereik
- a színezett homogenizátum fehérjetartalmát
- Köszönetnyilvánítás
- versengő érdekek
- kiegészítő adatok
anyagok és módszerek
biológiai minták – cékla, áfonya és vörösbor
cékla és áfonya homogenizátumokat készítettünk a kiegészítő anyagban leírtak szerint. A vörösbor nem igényelt fehérje extrakciót az U-2012 vizsgálat előtt.
kémiai reagensek
a nátrium-hipoklorit és a perklórsav (PCA) kivételével az összes kémiai reagenst Sigma vagy Sigma-Aldrich (St Louis, MO. USA). Nátrium-hipoklorit volt Acros Organic, New Jersey, USA. A PCA-t a BDH-tól (Anglia) szerezték be.
az U-1988 assay javításai
a karbonátról foszfátpufferre váltás 12,0 pH-n javította a reagens stabilitását és kis mértékben növelte az érzékenységet. Acetonitril került bevezetésre a mosószer okozta buborékok elkerülése érdekében. A NaOH helyettesítette a KOH-t, hogy elkerülje a kicsapódást a fehérje vizsgálatban. Ezenkívül a hatékonyságot fokozta a Lowry reagens különböző komponenseinek egyetlen reagenskeverékbe történő kombinálása.
az U-2012 vizsgálat
az U-2012 vizsgálat teljes részleteit a Kiegészítő anyag tartalmazza. Az 1. ábrán röviden összefoglalt protokoll leírja a vörösbor feldolgozását, valamint a cékla és áfonya homogenizátumait, és tartalmazza az U-1988 vizsgálat fejlesztéseit. Az U-2012 vizsgálatot feldolgozatlan, feldolgozott és fordított feldolgozású (H2O2 kezelés, majd TCA vagy PCA kicsapódás) fehérjékre alkalmazták. A vizsgálatokat BSA-n, karboanhidrázon, citokróm C-N, izocitrát-dehidrogenázon, lizozimon és tripszinen végeztük standard görbék és színes biológiai minták kifejlesztése céljából. A fehérjék meghatározását a biológiai mintákban a megfelelő standard görbékre történő kalibrálással végeztük.
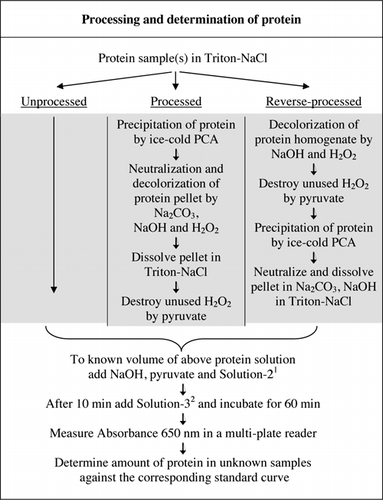
1a 2-es oldat réz-szulfátot (CuSO4.5h2o), Na-K-tartarátot, SDS-t és acetonitril-t tartalmazott 100 mM-es foszfátpufferben (pH 12,0). 2a 3-as oldathoz a Folin-Ciocalteu fenol reagensét hígítottuk 1:1 ionmentesített vízzel közvetlenül használat előtt.
a színinterferenciák becslését az U-2012-es vizsgálatban
a Színinterferenciát úgy határoztuk meg, hogy összehasonlítottuk a feldolgozott és a feldolgozatlan cékla -, áfonya-és vörösborminták abszorbenciáját Folin-reagens alkalmazásával és anélkül, az 1.ábrán leírtak szerint. Az arányt az interferencia mértékének meghatározására használtuk, ahol Abs1 a feldolgozatlan minták abszorbanciája Folin reagenssel; Abs2 a feldolgozatlan minták abszorbanciája Folin reagens nélkül; Abs3 a feldolgozott minták abszorbanciája Folin reagenssel; az Abs4 pedig a feldolgozott minták abszorbanciája Folin reagens nélkül.
a standard görbék kidolgozásához a mellékelt kiegészítő anyag receptek részében leírt standard görbét és paramétereit,
1b és 1c megoldást használtuk. A BSA koncentrációját és a megfelelő abszorbancia értékeket X-Y szórásgráf segítségével ábrázoltuk. Ennek a grafikonnak a formája (2.ábra) telítő választ mutat magasabb koncentrációkban, nagyon korlátozott kezdeti lineáris válasz mellett. Ez volt a korábban jelentett előnyös görbe forma (20). Kezdetben ezt exponenciális formában modellezték (19), de a későbbi vizsgálatok azt mutatták, hogy egy téglalap alakú hiperbola javította a választ, különösen alacsonyabb koncentrációk esetén. Ez utóbbi formát most szabványosították, és a következő háromparaméteres egyenletet használtuk az abszorbancia-fehérje koncentráció kapcsolat leírására:

Conc = fehérjekoncentráció,
a = abszorbancia Conc-nál,
A0 = abszorbancia nulla koncentrációnál,
AM= abszorbancia maximális koncentrációnál,
C50= abszorbanciát adó koncentráció
(AM + A0)/2.
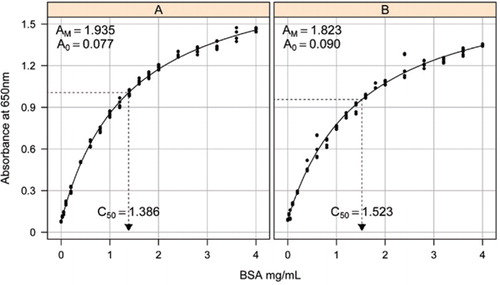
az abszorbancia (650 nm) és a BSA fehérje koncentrációja közötti standard görbe becslését három technikai ismétléssel végeztük (a) a feldolgozatlan fehérje vizsgálat (paraméterek A0= 0,077, Am=1,935, C50=1).386), és (B) a feldolgozott fehérje vizsgálat (paraméterek A0=0,90, Am=1,823, C50=1,523). A megfigyelt adatokhoz téglalap alakú hiperbola modellt (egyenletet ) illesztettünk. A paraméterbecsléseket a görbék a C50 becsléssel azonosítják az x tengelyen (pontozott fekete vonal).
az A0 paramétert kísérletileg határoztuk meg, míg az AM és a C50 értéket a Microsoft Excel Toolbox kiegészítő megoldó függvényével becsültük meg. Kísérleti paraméterkészletet használtunk a modellezett abszorbancia kiszámításához az egyes standard koncentrációknál (Conc) egyenlet segítségével . Ezután a megoldót arra utasították, hogy az AM és a C50 beállításával minimalizálja a standard készlet mért és modellezett abszorbanciája közötti maradék szórást.
megfigyeltük, hogy az abszorbancia és a koncentráció közötti kapcsolat nem lineáris görbét mutat a teljes koncentrációtartományban, ami valószínűleg a fényszórás egyik összetevőjének köszönhető, amely az abszorbancia mérésekor a fehérje koncentrációjának növekedésével növekszik. Coakley és James (20) alacsony abszorbancia mellett gyenge lineáris illeszkedést is jelentettek.
a homogenizátumok fehérjetartalmának kiszámítása
vizsgálatokat végeztek feldolgozott és feldolgozatlan vörösboron, valamint cékla és áfonya homogenizátumain. A BSA és más fehérjemintákat azonos módon kezeltük a megfelelő standard görbékhez az A0, AM és C50 értékek meghatározásához. Ezeket a paramétereket ezután arra használtuk, hogy a minta abszorbanciáját (A) az egyes homogenátumok fehérjekoncentrációjává alakítsuk át:

mivel az egyenletnek telítő formája van, az érzékenység csökken, mivel az abszorbancia (A), tehát a koncentráció növekszik. A fehérjebecslések hibái minimalizálhatók a homogenátok koncentrációjának a vizsgálatban történő beállításával, hogy azok ne lépjék túl a C50 értéket.
a homogenizátum Conc értékét ezután a következő képlet segítségével alakítottuk át szövetfehérje-koncentrációvá (szöveti Conc mg/g szövetben) :

ahol a homogenizált Conc-t (mg fehérje/mL-ben) a vizsgálat során korrigálták bármilyen előkoncentrációra vagy hígításra. Homogenizátum százalékos volt 100 g Szövet homogenizált, hogy a teljes térfogat 200 mL (a mi esetünkben 50%).
egy külön tanulmányban egy téglalap alakú hiperbola modellt szereltek fel a nem lineáris vegyes hatások (NLME) csomag (21) R (22) (2.ábra). A laboratóriumban egymástól függetlenül készített BSA-oldatot véletlenszerű hatásként modelleztük, közös A0, de eltérő AM és C50 együtthatóval. Ez modellezi a biológiai minták és a technikai vizsgálatok ismétlésének hierarchiáját.
eredmények és vita
fejlesztések az U-1988-as vizsgálatban
az U-1988-as és a Lowry-féle vizsgálat korlátai a karbonátalapú reagens instabilitása. A karbonát puffert (pH 11,4 2% = 188,7 mM) U-1988-ban 40 mM-es foszfáttal helyettesítettük 11,4-től 12,5-ig terjedő pH-értékeken. A 0,5 mg BSA/ mL-es és 1,0 mg BSA / mL-es BSA-val végzett FEHÉRJETESZTELÉS standard görbéiből kiinduló kezdeti lejtőket számítottuk ki. A foszfátpufferek 11,4-es pH-jával és optimális pH-jával rendelkező kezdeti lejtők 12,0 99 x10-6 és 197 x10-6 volt. A karbonát puffer meredeksége (pH 11,4) 162 x 10-6 volt.Mivel a meredekség értéke a vizsgálat érzékenységének közvetlen jele, foszfátpuffer (pH 12.0) a karbonát puffer helyettesítésére választották, 25% – kal növelve az érzékenységet.
nagyobb stabilitást értünk el a foszfátpuffer koncentrációjának 100 mM-re történő növelésével. a kapott foszfát/CuSO4/Na-K-tartarát oldat szobahőmérsékleten két hétig stabil volt, lényegesen hosszabb ideig, mint a karbonát/CuSO4/Na-K-tartarát oldat, amelyet naponta el kell készíteni a fehérje vizsgálat előtt. Minden jövőbeli kísérlethez 100 mM-es foszfátot (pH 12,0) használtunk a CuSO4/Na-K-tartarát oldat elkészítéséhez. Hisszük, hogy a karbonát foszfáttal történő cseréje növeli az U-2012 vizsgálat kényelmét.
a mosószer által indukált buborékok az abszorbancia mérésének fő hibaforrásává válnak, ha több lyukú lemezolvasót használnak (nem probléma a küvettákkal). Ezeket a buborékokat számos poláros oldószer (pl. aceton, acetonitril, etanol és metanol) hozzáadásával jelentősen csökkentettük. Ezek közül az oldószerek közül a legpolárisabb acetonitril (23) került kiválasztásra a hatékonysága miatt, és a 2.oldatba került (lásd a Kiegészítő anyag 1. ábrájának feliratát és receptjeit).
Foszfátpuffer, CuSO4, Na-K-tartarát, SDS és acetonitril külön-külön adhatók hozzá, és az adagolás sorrendje nem befolyásolja a kapott abszorbanciát. Az előkevert oldat használata azonban tovább növeli a kényelmet, különösen akkor, ha nagy számú mintát kell vizsgálni. Ezért a vizsgálati keverék ezen komponenseit 2-es oldatba csoportosítottuk (1.ábra). Az eredeti Lowry-vizsgálat (6) esetében ilyen előkevert oldat nem volt megvalósítható a karbonátoldat instabilitása miatt. A 3-as oldat 2-es oldatba való beillesztésének kísérlete a kék szín fejlődésének drámai csökkenését eredményezte, ezért nem vették figyelembe tovább.
fehérje becslés színes biológiai mintákban
fehérje extrakció
a cékla és áfonya fehérjéit Triton X-100-NaCl oldatban extraháltuk enyhe homogenizációval. Az ilyen homogenizátumok megtartják enzimaktivitásukat (15). Ez az extrakció nem volt szükséges a vörösbor esetében.
interferáló anyagok eltávolítása
színes minták esetében el kell távolítani a mintában rejlő színből és más nem fehérjeanyagokból eredő interferenciát, amelyek a kolorimetriás fehérje-vizsgálat előtt reagálnak a fehérjereagensekkel. Az U-2012 újdonsága egy színtelenítő protokoll kidolgozása, amely kompatibilis a kolorimetriás fehérje vizsgálattal.
a színes pigmentek nátrium-hipoklorit vagy H2O2 általi Színtelenítését, valamint a fehérjék PCA vagy TCA általi szelektív kicsapását mérlegelték a zavaró anyagok eltávolítása céljából. A nátrium-hipokloritot, a H2O2-t, a TCA-t és a PCA-t értékelték az U-2012 vizsgálattal való kompatibilitásuk szempontjából, BSA-t használva tesztfehérjeként. A nátrium-hipoklorit és a H2O2 között csak a H2O2 volt kompatibilis, mivel hipoklorit jelenlétében Csapadék képződött. A TCA vagy PCA által kicsapott fehérjék u-2012-vel vizsgálhatók a pelletben lévő maradék sav megfelelő semlegesítése után. Beszámoltak a PCA fölényéről a TCA-val szemben a fehérje kicsapódásában (24,25). Ezzel szemben összehasonlító értékelésünkben hasonló C50 értékeket tártunk fel a PCA (1.395) és a TCA (1.400) esetében. A PCA-t részesítettük előnyben, mivel előre elkészített oldatként (70% v/v) könnyen elérhető, ezért könnyen hígítható a kívánt szilárdságra. A TCA higroszkópos szilárd anyag, amelyet változó víztartalma miatt nehéz pontosan megmérni.
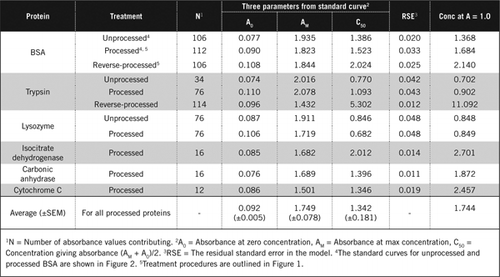
a PCA és a H2O2 kombinálásának két lehetséges módja van. A “feldolgozott” fehérjék esetében a PCA-kezelést H2O2-kezelés követte, a “fordított feldolgozású” fehérjék esetében pedig a H2O2-kezelés előzte meg a PCA kicsapódását. A feldolgozott fehérje használatának előnyei a felülúszóban lévő számos zavaró anyag eltávolítása és a proteolitikus enzimek lehetséges inaktiválása voltak a minta előkészítése során. Ezt a feldolgozott és fordított tripszin és BSA vizsgálatok igazolták (lásd 1.táblázat). Csak feldolgozott mintákat használtunk a színes biológiai minták tényleges fehérjetartalmának meghatározására
mind a PCA, mind a H2O2 színes minták kezelése szükséges volt az interferencia megszüntetéséhez az U-2012 vizsgálatban. A színes minták savas kicsapódása önmagában nem távolította el teljesen az interferenciát. Az összes színes mintával néhány színt eldobtak a felülúszóban, de a pelleteket is színezték. A pellet színét H2O2 kezeléssel távolították el. Lúgos körülményekre volt szükség mind a H2O2 (26) általi hatékony színtelenítéshez, mind a Folin reagens általi színfejlődéshez a fehérjeszint helyes mérésének biztosítása érdekében. Bár mind a NaOH, mind a KOH biztosítani tudta a szükséges lúgosságot, csak a NaOH volt kompatibilis az U-2012 vizsgálattal. Koh jelenlétében Csapadék képződött. Pelletekben a PCA-t Na2CO3 és NaOH (27) alkalmazásával semlegesítettük. A vizsgálat során további NaOH-t adtunk hozzá; az optimalizált térfogat 50-70 6L között volt (rutinszerűen 60 6L-t használtunk); lásd az 1.ábrát.
céklát, áfonyát és vörösbort színtelenítettünk 15 60% – os H2O2-val, 0,5 és 2 órát 50 oc-on és szobahőmérsékleten. Húsz mikroliter 30% H2O2 1 órán át 50cc-n, hogy megbirkózzanak az erősebb színű mintákkal. Az olyan anyagok oxidációja, mint a cukrok, amelyek H2O2-vel kötődnek a fehérjékhez 50 Kb kritikusnak tűnik, mivel a szobahőmérséklet feldolgozása túlbecsüli a fehérjetartalmat. A cékla esetében az 50 db C feldolgozás a látszólagos fehérjebecslést a feldolgozatlan 14% – ára csökkentette, míg a szobahőmérséklet feldolgozása csak a felére csökkentette ezt a becslést.
a hidrogén-peroxid-kezelés után végzett kolorimetriás vizsgálatokból nyilvánvaló volt, hogy a színtelenítés során bizonyos H2O2-t nem használtunk. Egy ilyen mintában a Lowry-vizsgálat végszíne részben megsemmisült. Ezért a maradék hidrogén-peroxidot el kellett pusztítani a fehérje vizsgálat előtt. Az U-2012 vizsgálatban két H2O2 forrás található; a színtelenítéshez hozzáadott H2O2, a Triton X-100-ban pedig szennyező anyagként jelen lévő H2O2 (0,22%, Termékinformáció: Triton X-100, www.sigmaaldrich.com). a hidrogén-peroxidot általában a kataláz enzim bontja le. A fehérje vizsgálat magas pH-ja azonban inaktiválná az ismert katalázokat. Ezenkívül a kataláz hozzáadása extra fehérje hozzáadásához vezetne. A H2O2 kémiai megsemmisítését választottuk piruvát alkalmazásával (28). A piruvát-H2O2 kölcsönhatási egyenlet kémiája jól megalapozott (28,29). A piruvát szobahőmérsékleten elpusztítja a H2O2-t a következő reakció szerint:

a maradék H2O2-t a pellet szuszpenzióban 0,9 M piruváttal (1,5 x H2O2 koncentráció) 0,5 órán át szobahőmérsékleten kezeltük. A Triton X-100-ban a szennyező H2O2 ellensúlyozására extra piruvátot is hozzáadtunk a fehérje vizsgálathoz (1.ábra). A piruvát hozzáadása alacsonyabb abszorbanciát adott egy nem fehérje vak számára . Javasoljuk, hogy a Triton X-100 peroxid-szennyezője reagáljon az acetonitril-2 oldatban, kissé nagyobb abszorpciót biztosítva.
a színes biológiai mintákhoz kapcsolódó színinterferenciát nem lehet egyszerűen figyelembe venni egy fehérje vizsgálat futtatásával a Folin reagens hiányában. A számított arányok (Abs1-Abs2)/(Abs3-Abs4) azt mutatták, hogy a mintaszín interferenciája a vörösbor ( = 40), az áfonya ( = 6) és a cékla ( = 2) esetében volt a legnagyobb. Ez az interferencia a valódi fehérjeszintek abnormálisan magas becsléseire vezethető vissza; például a fehérje koncentrációja feldolgozatlan és feldolgozott cékla homogenátumok felhasználásával(20,21 versus 2,89 mg fehérje / g Szövet). A színinterferencia mellett a vörösbor, valamint a cékla és áfonya homogenizátumai valószínűleg olyan anyagokat tartalmaznak, amelyek az U-2012 vizsgálatban reagálnak a Folin reagensével (pl.g., kis peptidek és komplex cukrok). Ezeket úgy távolítottuk el, hogy a fehérjéket jéghideg PCA-val szelektíven kicsaptuk 5% – os végső koncentrációban (1.ábra).
Standard görbék és paramétereik
a feldolgozatlan és feldolgozott BSA Standard görbéit a 2.ábra mutatja. A származtatott paraméterek (A0, AM és C50) szintén szerepelnek az 1.táblázatban a BSA és más fehérjék esetében.
az eredmények azt mutatják, hogy a modellben a maradék standard hiba alacsony (0,012-0,048), ami azt jelzi, hogy az adatok jobban illeszkednek a téglalap alakú hiperbola trendhez. A különböző fehérjék és feldolgozásuk közötti információk összehasonlításához a paramétereket az abszorbancia = 1,0 koncentrációjára konvertáltuk 650 nm-en (az 1. táblázat jobb oldali oszlopa).
ezek az eredmények azt mutatják, hogy a feldolgozott mintákban a fehérjeveszteség (a feldolgozatlan fehérjével összehasonlítva) kisebb volt, mint a fordított feldolgozású mintákban. Ez a veszteség a tripszin esetében nyilvánvalóbb volt, és a fordított feldolgozás során végzett auto-katalitikus aktivitásával magyarázható. Javasoljuk, hogy a feldolgozott protokollt (Kiegészítő anyag) csak olyan biológiai minták esetében kövessék, amelyek valószínűleg proteolitikus enzimeket tartalmaznak.
az eredeti Lowry-vizsgálatban (6) és annak módosított változatában, az U-1988-ban (15) a fehérje mennyiségi meghatározásához a standard görbének csak az abszorbanciának a fehérjemennyiséggel szembeni ábrázolásával kapott lineáris részét használtuk. Az U-2012 vizsgálatban az adatokat hatékonyabban használjuk fel egy téglalap alakú hiperbola-egyenlet illesztésével, amint azt az anyagok és módszerek részben leírtuk Coakley és James (20) szerint.
a színezett homogenizátum fehérjetartalmát
az ismeretlen mintákban a Fehérjekoncentrációkat egyenlet alapján és a feldolgozott BSA standard és az 1.táblázatban felsorolt összes feldolgozott fehérje átlaga alapján számítottuk ki. Ez utóbbi közelebb áll a fehérjék keverékét tartalmazó biológiai minták valódi becsléséhez. Az áfonya és a cékla fehérjemennyiségét a vörösborhoz viszonyítva körülbelül 60, illetve 230-szorosra becsültük (2. táblázat).
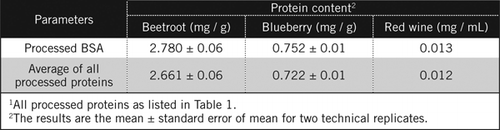
a BSA-hoz hasonlóan a vörösbort és a cékla és áfonya 50% – os homogenizációját PCA kicsapással és H2O2-vel történő színtelenítéssel dolgozták fel (1.ábra). Ebben a szakaszban a biológiai mintákat a vörösborra 40-szer, a céklára és az áfonyára 4-szer koncentráltuk. Hasonlóképpen BSA (2 mg/mL) is koncentrált 4-szer 8 mg/mL. A színes minták abszorbanciáját, amely közel volt a C50 (feldolgozatlan BSA) abszorbanciájához, a fehérjetartalom kiszámításához használtuk, az AND egyenletben leírtak szerint .
összefoglalva, az U-2012 vizsgálat stabil reagenseket alkalmazott, jobb érzékenységet biztosított (még a színtelen biológiai minták esetében is), és legyőzte a színes biológiai minták szín okozta interferenciáját. Az U-2012 vizsgálat nem korlátozódik a fehérjekoncentráció és az abszorbancia közötti válasz lineáris részére, és a nemlineáris régióban az adatok hatékonyabb felhasználását teszi lehetővé a szabványokhoz illesztett téglalap alakú hiperbolikus görbe modell segítségével, a Microsoft Excel egyszerű eljárásaival.
Köszönetnyilvánítás
a szerzők elismerik az Új-zélandi kutatási, tudományos és technológiai Alapítványt pénzügyi támogatásért (c06x0809).
versengő érdekek
a szerzők kijelentik, hogy nincsenek versengő érdekek.
kiegészítő adatok
a cikkhez mellékelt kiegészítő adatok megtekintéséhez kérjük, látogasson el a folyóirat weboldalára: www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R. S. és P. L. Pedersen. 1985. Mikrogramm mennyiségű fehérje meghatározása milligramm lipidszint jelenlétében amido black 10B-vel. Anális. Biochem. 150:97–104.Crossref, Medline, CAS, Google Tudós
- 2. Gornall, A. G., C. J. Bardawill és M. M. David. 1949. A szérumfehérjék meghatározása biuret reakcióval. J. Biol. Kémia. 177:751–766.Medline, CAS, Google Tudós
- 3. Kovács, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke et al.. 1985. A fehérje mérése bicinchoninsavval. Anális. Biochem. 150:76–85.Crossref, Medline, CAS, Google Tudós
- 4. Bradford, M. M. 1976. Gyors és érzékeny módszer a fehérje mikrogramm mennyiségének meghatározására a fehérje-festék kötés elve alapján. Anális. Biochem. 72:248–254.Crossref, Medline, CAS, Google Tudós
- 5. Zor, T. és Z. Selinger. 1996. A Bradford protein assay linearizációja növeli érzékenységét: elméleti és kísérleti vizsgálatok. Anális. Biochem. 236:302–308.Crossref, Medline, CAS, Google Tudós
- 6. Lowry, O. H., N. J. Rosbrough, A. L. Farr és R. J. Randall. 1951. Fehérjemérés Folin fenol reagenssel. J. Biol. Kémia. 193:265–275.Medline, CAS, Google Tudós
- 7. Peterson, G. L. 1979. Lowry, Rosebrough, Farr és Randall Folin fenol fehérje mennyiségi meghatározási módszerének áttekintése. Anális. Biochem. 100:201–220.Crossref, Medline, CAS, Google Tudós
- 8. Sapan, C. V., R. L. Lundablad és N. C. Price. 1999. Kolorimetriás fehérje vizsgálati technikák. Biotechnol. Appl. Biochem. 29:99–108.Medline, CAS, Google Tudós
- 9. Okutucu, B., A. D., Á., Á., Á., Á., Á., Á., Á., Á., Á. Habib, és F. Z. 2007. A teljes plazmafehérje-koncentráció meghatározására szolgáló öt módszer összehasonlítása. J. Biochem. Biophys. Módszerek 70: 709-711.Crossref, Medline, CAS, Google Tudós
- 10. Kresge, N., R. D. Simoni és R. L. Hill. 2005. A kiadói történelem legtöbbet idézett cikke: fehérje meghatározása Oliver H. Lowry. J. Biol. Kémia. 25:280.Google Tudós
- 11. Everette, J. D., Q. M. Bryant, A. M. Green, Y. A. Abbey, G. W. Wangila és R. B. Walker. 2010. A különböző vegyületosztályok reaktivitásának alapos vizsgálata a Folin-Ciocalteu reagens felé. J. Agric. Élelmiszer-Kémia. 58:8139–8144.Crossref, Medline, CAS, Google Tudós
- 12. Eichberg, J. és L. C. Mokrasch. 1969. Az oxidált lipidek interferenciája a fehérje meghatározásában a Lowry eljárással. Anális. Biochem. 30:386–390.Crossref, Medline, CAS, Google Tudós
- 13. Dulley, J. R. és P. A. gyászolnak. 1975. Egyszerű technika a mosószerek interferenciájának kiküszöbölésére a fehérje meghatározásának Lowry módszerében. Anális. Biochem. 64:136–141.Crossref, Medline, CAS, Google Tudós
- 14. Brillouet, J.-M., M.-P. Belleville és M. Moutounet. 1991. Lehetséges fehérje-poliszacharid komplexek vörösborokban. Am. J. Enol. Vitic. 42:150–152.CAS, Google Tudós
- 15. Upreti, G. C., R. A. Ratcliff és P. C. Riches. 1988. Fehérje becslés magas lipidszintet tartalmazó szövetekben: a fehérje meghatározásának Lowry-módszerének módosításai. Anális. Biochem. 168:421–427.Crossref, Medline, CAS, Google Tudós
- 16. Upreti, G. C., C. Davis és J. Oliver. 1991. Biológiai szövetek reprezentatív homogenizátumainak előállítása: a só hatása a fehérje extrakcióra. Anális. Biochem. 198:298–301.Crossref, Medline, CAS, Google Tudós
- 17. Smith, M. R., M. H. Penner, S. E. Bennett és A. T. Bakalinsky. 2011. A Pinot Noir vörösborra alkalmazott összes fehérje mennyiségi kolorimetriás vizsgálata. J. Agric. Élelmiszer-Kémia. 59:6871–6876.Crossref, Medline, CAS, Google Tudós
- 18. Wigand, P., S. Tenzer, H. Schild és H. Decker. 2009. A vörösbor fehérjeösszetételének elemzése A Ros-és Fehérborokkal összehasonlítva elektroforézissel és nagynyomású folyadékkromatográfiás tömegspektrometriával (HPLC-MS). J. Agric. Élelmiszer-Kémia. 57:4328–4333.Crossref, Medline, CAS, Google Tudós
- 19. Upreti, G. C., Y. Wang, A. Sharrock, N. Feisst, M. Davy és B. Jordan. 2009. Stabil és érzékeny fehérje teszt (U-2009 módosított teszt) színes biológiai mintákra. Kombiol., Új-Zéland Végleges Programja 2009. December. Canterbury Egyetem, Christchurch, Új-Zéland.Google Tudós
- 20. Coakley, W. T. és C. J. James. 1978. Egy egyszerű lineáris transzformáció a Folin-Lowry fehérje kalibrációs görbéhez 1,0 mg/mL-re. Anális. Biochem. 85:90–97.Crossref, Medline, CAS, Google Tudós
- 21. Pinheiro, J. C. és D. M. Bates. 2000. Vegyes hatású modellek S és S-PLUS, statisztikák és Számítástechnikai sorozatokban. Springer-Verlag, New York, NY.Google Tudós
- 22. R Development Core Csapat. 2009. R: a statisztikai Számítástechnika nyelve és környezete. R statisztikai számítástechnikai Alapítvány, Bécs, Ausztria, ISBN 3-900051-07-0, URL http://www.R-project.org.Google tudós
- 23. Khachik, F., G. R. Beecher, J. T. Vanderslice és G. Barázda. 1988. Folyadékkromatográfiás leletek és csúcstorzulás: Minta-oldószer kölcsönhatás a karotinoidok elválasztásában. Anális. Kémia. 60:807–811.Crossref, Medline, CAS, Google Tudós
- 24. Cernik, A. A. 1970. Az etilén-diamin-Tetra-ecetsavval kelátált ólom meghatározása a vérben a fehérje perklórsavval történő kicsapódása után. Brit. J. Industry Med. 27:40–42.Medline, CAS, Google Tudós
- 25. Moughan, P. J., A. J. Darragh, W. C. Smith és C. A. Butts. 1990. Perklór – és triklór-ecetsavak, mint a patkány endogén ileális digestájában a fehérje kicsapódása. J. Sci. Élelmiszer Agric. 52:13–21.Crossref, CAS, Google Tudós
- 26. Galb, Z. M. és L. J. CS. 1983. A hidrogén-peroxid alkáli-indukált bomlása. J. Kémia. Soc., Dalton Trans. 11:2353–2357.Crossref, Google Tudós
- 27. Scopes, R. K. 1988. Protein Tisztítás: elvek és gyakorlat, második Ed. Springer-Verlag New York Inc., New York, NY.Google Tudós
- 28. Upreti, G. C., K. Jensen, R. Munday, D. M. Duganzich, R. Vishwanath és J. F. Smith. 1998. Tanulmányok az aromás aminosav-oxidáz aktivitásról a ram spermatozoidokban: a piruvát szerepe antioxidánsként. Anim. Fedd. Sci. 51:275–287.Crossref, Medline, CAS, Google Tudós
- 29. Holleman, M. A. F. 1904. Megjegyzés: sur l ‘action de l’ eau oxygenee sur les savak-cetoniques et sur les dicetones 1.2. Pihenő. Trav. Chim. Pays-Bas Belg. 23:169–172.Crossref, CAS, Google Tudós