Introducción
La distrofia neuroaxonal infantil (INAD) es una enfermedad neurodegenerativa rara autosómica recesiva de frecuencia desconocida. La aparición de los síntomas generalmente ocurre entre los 6 meses y los 3 años de edad. Antes de ese momento, los bebés se desarrollan normalmente. El primer síntoma de INAD puede ser la desaceleración del logro de los hitos normales del desarrollo o la regresión en los hitos del desarrollo (Ramanadham et al., 2015). La hipotonía del tronco, el estrabismo y el nistagmo son síntomas tempranos de la enfermedad (Gregory et al., 2017). La progresión de la enfermedad es rápida y, a medida que avanza, se pierden más habilidades adquiridas. Los músculos pronto se vuelven hipotónicos y más tarde espásticos (Levi y Finazzi, 2014). Con el tiempo, se pierde todo el control muscular voluntario. La debilidad muscular también puede provocar dificultades para alimentarse y respirar. Además del nistagmo, algunos niños experimentan pérdida de la visión. Las funciones cognitivas se pierden gradualmente y se desarrolla demencia. La esperanza de vida es generalmente de 5 a 10 años (Gregory et al., 1993; Jardim et al., 2004; Macauley y Sands, 2009).
Patología molecular
La distrofia Neuroaxonal infantil pertenece a una familia de trastornos neurodegenerativos que incluye la distrofia neuroaxonal atípica de inicio tardío (ANAD) y el complejo de Parkinsonismo distónico (DPC). La mayoría de los casos de INAD están asociados con mutaciones homocigotas o heterocigotas compuestas en el gen PLA2G6 que afectan la actividad catalítica de su producto proteico (Engel et al., 2010). El gen PLA2G6 codifica un grupo a través de la proteína fosfolipasa A2 independiente del calcio (PLA2G6 o iPLA2ß, ∼85/88 kDa) con una lipasa y siete dominios que contienen repetición de anquirina (Tang et al., 1997). PLA2G6 hidroliza la cadena acilo sn-2 de fosfolípidos, generando ácidos grasos libres y lisofosfolípidos.
Los fosfolípidos en la membrana interna de las mitocondrias son ricos en ácidos grasos insaturados en la posición sn-2, particularmente cardiolipina (Seleznev et al., 2006). Estos ácidos grasos insaturados son particularmente vulnerables a las abundantes especies reactivas de oxígeno producidas por las mitocondrias (Murphy, 2009), lo que resulta en fosfolípidos peroxidados en la membrana interna de las mitocondrias. PLA2G6 se localiza en las mitocondrias (Williams y Gottlieb, 2002; Liou et al., 2005) consistente con una mayor demanda de hidrólisis de los ácidos grasos peroxidados en la posición sn-2 de los fosfolípidos que conduce a fosfolípidos remodelados (Balsinde et al., 1995; Zhao et al., 2010). Cuando PLA2G6 es defectuoso, la integridad de la membrana interna de la mitocondria se daña. PLA2G6 también se localiza en el axón (Ong et al., 2005; Seleznev et al., 2006), lo que indica un aumento de la demanda localizada de remodelado de fosfolípidos allí también. La manifestación de dicha acumulación en el cerebro es exclusiva de áreas clave del cerebro, como los ganglios basales, que dieron lugar a varios nombres para la misma patogénesis molecular subyacente que involucra PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
El análisis ultraestructural de neuronas en ratones knockout PLA2G6 es consistente con esta patología molecular. Se han observado mitocondrias con cristales tubulares y ramificados, mitocondrias con cristales degenerados, axones con colapso del citoesqueleto y pérdida parcial de la membrana en los terminales de los axones (Beck et al., 2011). A nivel microscópico, estas características aparecen como inflamaciones axonales y cuerpos esferoides en terminales pre-sinápticas (Figura 1) en el sistema nervioso central o periférico.FIGURA
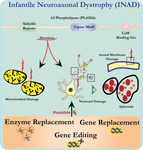
Gráfico 1 La distrofia neuroaxonal infantil (INAD) es un trastorno neurodegenerativo relacionado con mutaciones en el gen PLA2G6. Varias mutaciones de PLA2G6 conducen a una fosfolipasa A2 disfuncional que conduce a defectos de la membrana mitocondrial y axonal. Estos defectos causan daño neuronal visualizado como hinchazones axonales y acumulación de esferoides pre-sinápticos. El reemplazo enzimático para restaurar funciones, el reemplazo de genes o la edición para corregir el PLA2G6 defectuoso son estrategias terapéuticas propuestas.
Diagnóstico molecular y Empoderamiento de Pacientes con Enfermedades Raras
Aparte de características clínicas, electrofisiológicas y de imagen específicas, antes de la disponibilidad de secuenciación de próxima generación, las biopsias de piel que mostraban hinchazones axonales y cuerpos esferoides en terminales pre-sinápticos en el sistema nervioso central o periférico fueron los criterios diagnósticos para la confirmación del INAD (Gregory et al., 2017; Iodice et al., 2017). A menudo, se requerían múltiples biopsias para confirmar el diagnóstico. Por lo general, las familias esperaron muchos años para recibir un diagnóstico. Con el costo decreciente de la secuenciación de genes y genomas, la disponibilidad de pruebas de panel de genes específicos en laboratorios de diagnóstico para enfermedades neurológicas no diagnosticadas y la creciente conciencia de los médicos sobre la disponibilidad de diagnósticos genéticos, las familias están recibiendo el diagnóstico más rápidamente; a veces dentro de un año de la aparición del primer síntoma. Los niños de estas familias todavía son pequeños y las familias están motivadas a asociarse con científicos para encontrar un tratamiento para la enfermedad de sus hijos. Para financiar el proceso de investigación, un grupo de padres de pacientes con INAD formó la Fundación INADcure. La fundación ha recaudado fondos sustanciales para la investigación y se ha asociado con el Instituto de Genómica Rara para guiarlos en la concesión de becas de investigación. Curiosamente, un análisis genético de 22 familias indias con INAD, ANAD y DPC de 2016 encontró que 10/22 familias (45,45%) carecían de mutaciones en la región codificadora del gen PLA2G6 (Kapoor et al., 2016). La falta de identificación de mutaciones perjudiciales en la región codificante de PLA2G6 pone de relieve que los futuros esfuerzos de diagnóstico molecular requerirían la secuenciación de genes completos para identificar mutaciones en las regiones intrónicas y reguladoras del gen PLA2G6 en pacientes afectados por INAD. Además, en los casos de DAD en los que no se observan mutaciones asociadas a PLA2G6, se sugiere que la causa de la enfermedad podría deberse a mutaciones en genes distintos de PLA2G, que deben explorarse.
Posibles terapias
La mayoría de las terapias que se consideran para una enfermedad rara como la INAD que involucra una enzima defectuosa son el reemplazo de enzimas, el reemplazo de genes o la corrección de genes. Cuando una deficiencia enzimática es causada por un defecto genético recesivo, se asume que el reemplazo o la suplementación enzimática puede corregir el problema (Smith et al., 2012; Yu-Wai-Man, 2016). Sin embargo, los experimentos para probar que las terapias que proporcionan el gen o enzima correctos rescatarán el fenotipo INAD aún no se han realizado y probado.
Terapia de Reemplazo Enzimático (ERT)
Dado que el cerebro es el órgano principal afectado en el INAD, la terapia de reemplazo enzimático para el INAD probablemente requeriría una infusión de la enzima en el cerebro. En 2017, Biomarin recibió la aprobación de la FDA para tripeptidil peptidasa 1 (cerliponasa alfa) como tratamiento para la causa subyacente de la enfermedad de Batten, la deficiencia de tripeptidil peptidasa (TPP1), una enzima lisosómica (Administración de Medicamentos Alimentarios de los Estados Unidos, 2017). Tripeptidil peptidasa 1 (cerliponasa alfa; ∼59 kDa) es la primera ERT que se administra directamente en el líquido cefalorraquídeo (LCR) del cerebro. Otros fármacos ERT que se pueden administrar en el LCR se encuentran en ensayos clínicos (Jardim et al., 2004; Macauley y Sands, 2009).
La eficacia de la tripeptidil peptidasa 1 (cerliponasa alfa) en la capacidad de caminar de los pacientes con enfermedad de Batten demuestra que la terapia de reemplazo enzimático para enfermedades que afectan al cerebro es teóricamente posible. Dirigir la enzima de reemplazo a las mitocondrias para el INAD será más difícil que dirigir la enzima lisosómica, que ya había tenido éxito con la ERT IV durante mucho tiempo, como en el caso de la enfermedad de Gaucher. Por lo tanto, todavía hay muchos problemas biológicos específicos de INAD que deben resolverse:
* ¿La terapia para INAD necesita mecanismos que puedan transportar PLA2G6 a la célula?
* ¿Qué mecanismo se puede utilizar para localizar PLA2G6 a las mitocondrias?
* ¿Puede una PLA2G6 parcial rescatar su deficiencia o se requiere una proteína completa?
* ¿Cuál es un disolvente apropiado para la administración del producto del gen PLA2G6 en el líquido cefalorraquídeo?
• Si la enzima PLA2G6 no es 100% pura, ¿cuáles son sus contaminantes y son seguros?
* ¿Habrá alguna complicación intravascular generalizada debido a la infusión de la enzima PLA2G6 en el SNC? ¿La dosis de infusión tiene alguna relevancia para tales complicaciones?
Un problema adicional es que la terapia de reemplazo enzimático requiere la colocación inicial de un catéter intracerebroventricular y perfusiones frecuentes. La colocación del catéter requiere anestesia y las infusiones pueden requerir anestesia dependiendo de la cooperación de los pacientes. Abordar las preocupaciones de los neurólogos pediátricos y los padres de niños con INAD con respecto al uso de anestesia en pacientes con INAD con información adecuada requerirá un mayor esfuerzo.
Terapia génica / Reemplazo Génico
El gen PLA2G humano es de alrededor de 70 kb con 17 exones y 2 exones alternativos. La transcripción de codificación de proteínas más larga, sin embargo, es de solo 3,3 kb y la secuencia de codificación de proteínas es de poco más de 2,4 kb, que se puede empaquetar fácilmente en una carga de vectores virales. Primero exploraremos otro trastorno de almacenamiento lisosomal en el SNC que tiene datos preclínicos para luego compararlos con INAD. La falta de datos preclínicos de TRE en el INAD es una brecha de conocimiento significativa en el campo, por lo que es crucial que observemos otros modelos preclínicos de trastornos del SNC que están siendo sometidos a estudios de terapia génica. Anteriormente, un estudio preclínico en ratones abordó la terapia génica para el trastorno de almacenamiento lisosomal mucopolisacaridosis tipo IIIA (MPS-IIIA) empaquetando la versión correcta del gen de la sulfamidasa dentro de un vector viral (Sorrentino et al., 2013). El estudio aprovechó la creciente comprensión de la barrera hematoencefálica (BBB), que regula activamente el transporte de moléculas grandes de la sangre al SNC mediante un proceso llamado transcitosis (Pardridge, 2005b; Sorrentino y Fraldi, 2016).
La transcitosis implica la endocitosis de ligandos en el lado luminal, mediada por receptores específicos de ligandos (por ejemplo, receptor de insulina, receptor de transferrina y receptor de lipoproteínas de baja densidad, etc.).,) enriquecido en el endotelio capilar (Pardridge, 2005a). A esto le sigue el movimiento de la carga endocitosada a través del citoplasma del endotelio y, finalmente, la exocitosis en el lado abluminal (cerebro), entregando así efectivamente la carga a través de la BBB (Pardridge, 2002). El estudio preclínico MPS-IIIA fabricó una proteína sulfamidasa quimérica que contenía un dominio de unión a BBB de la apolipoproteína B para facilitar la absorción por el endotelio y también un péptido señal de la iduronato-2-sulfatasa para ayudar a una exocitosis eficiente hacia el lado abluminal de la BBB (Sorrentino et al., 2013). A continuación, se cargó un vector viral que codificaba una sulfamidasa quimérica en un serotipo 2/8 del virus adeno asociado (AAV) dirigido al hígado (Sorrentino y Fraldi, 2016). Por lo tanto, el hígado sirvió como una fábrica interna que proporcionó un suministro constante de sulfamidasa quimérica, lo que resultó en un aumento del 10-15% en la actividad de la sulfamidasa cerebral incluso 7 meses después de la terapia génica hepática. Este aumento en los niveles de actividad de la sulfamidasa cerebral condujo a una mejora cuantificable en los resultados de la patología cerebral y el comportamiento en el modelo de ratón de MPS-IIIA (Sorrentino et al., 2013).
Si se realiza un estudio similar para el INAD, respondería a varias preguntas críticas sobre el reemplazo enzimático para el tratamiento con INAD. Alternativamente, la administración intravascular o intra-LCR de productos de terapia génica basados en AAV9 puede dirigirse directamente al SNC (Bey et al., 2017; Roca et al., 2017). Sin embargo, las múltiples mutaciones que los pacientes con INAD tienen en su gen PLAG26 ofrecen desafíos únicos a la terapia génica. Aunque el tamaño del gen PLA2G6 de 2-3 kb no debería suponer un problema para su inserción en un vector viral, sin embargo, las complicaciones reguladoras de la corrección de PLA2G6 son difíciles de predecir. Las complicaciones regulatorias se vuelven incontrolables, especialmente si la terapia génica no puede localizarse adecuadamente dentro de los tejidos diana. La terapia de reemplazo enzimático también puede ser un problema si algunos productos mutantes resultan ser negativos para el PLAG26 de tipo salvaje.
La mayoría de los desafíos de las posibles terapias para la INAD provienen del hecho de que es una enfermedad ultra rara. Sin embargo, el hecho de que el INAD tenga una etiología genética conocida proporciona vías para posibles terapias. Además, cualquier terapia exitosa para INAD recibirá el estatus de medicamento huérfano y toda la protección que recibe, porque INAD es una enfermedad rara. Por lo tanto, a pesar de todos los desafíos mencionados, el estatus de medicamento huérfano proporciona un fuerte incentivo para los investigadores de enfermedades raras y la industria de la biotecnología, por no mencionar cuando la causa genética ya se conoce.
Edición de genes/Bases
A partir de 2017, se han observado al menos 277 mutaciones sin sentido en el gen PLA2G6 humano (Lek et al., 2016). Solo una pequeña proporción de mutaciones PLA2G6 incluye cambios de marco, indels, mutaciones sin sentido y mutaciones en los sitios de empalme (Morgan et al., 2006). Por lo tanto, corregir el gen en la población de células diana o corregir las bases de ADN mutadas es una vía terapéutica atractiva. A lo largo de los años, se han desarrollado varias herramientas para la edición de genes, y la edición del genoma basada en CRISPR/Cas9 está siendo aclamada como una tecnología innovadora con un ensayo clínico planeado para 2018. Estas tecnologías utilizan una proteína de unión al ADN que también puede escindir la hebra de una manera específica para hacer espacio para la inserción de una nueva secuencia de ADN o la corrección de la base del ADN perjudicial (LaFountaine et al., 2015). La tecnología CRISPR/Cas9 ya ha dado resultados terapéuticos asombrosos en varios modelos de enfermedades preclínicas (por ejemplo, distrofia muscular de Duchene, enfermedades metabólicas hepáticas, etc.).,) (Dai et al., 2016). Además, una comprensión creciente de la variación humana está planteando nuevos desafíos a la terapia génica e impulsando la innovación hacia una verdadera personalización de la edición génica (Lessard et al., 2017; Scott y Zhang, 2017). Las nuevas tecnologías como vSLENDR, un virus AAV, y la tecnología mediada por CRISPR/Cas9 para reemplazar genes defectuosos en neuronas y otras células del sistema nervioso están empujando las tecnologías de edición de genes a nuevas fronteras (Nishiyama et al., 2017).
Conclusión
La distrofia neuroaxonal infantil es una enfermedad neurodegenerativa grave con cierta morbilidad y mortalidad. Esta enfermedad rara ofrece una oportunidad emocionante para revalidar los modos disponibles de terapia de próxima generación y generar otros más nuevos. Se espera que la congruencia emergente con los criterios de diagnóstico clínico del INAD impulse el diagnóstico molecular mejorado. Se espera que este progreso nos lleve a desarrollar terapias asequibles que proporcionen una mejora cuantificable en la calidad de vida de los pacientes con INAD y retarden o mejoren la progresión de la enfermedad. Hemos destacado algunas historias de éxito en la enfermedad de Batten y las mucopolisacaridosis que nos proporcionan la inspiración para hacer las preguntas correctas para hacer realidad la terapia INAD. Además, el crecimiento de enfoques virales y no virales para la edición de genes basados en CRISPR/Cas9 también debería abrir nuevas vías terapéuticas para el INAD.
Contribuciones de autores
PB y FA prepararon un borrador preliminar. SR y AC editaron y agregaron más secciones al manuscrito. DF, AP y LP editaron el manuscrito.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. A. (1995). La inhibición de la fosfolipasa A2 independiente del calcio impide la incorporación de ácido araquidónico y la remodelación de fosfolípidos en macrófagos P388D1. Proc. Natl. Acad. Sci. U. S. A. 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Focalización eficiente del SNC en ratones adultos mediante infusión intratecal de AAV9-GFP monocatenario para terapia génica de trastornos neurológicos. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 para terapia génica in vivo: promesa y obstáculos. Mol. Allí. Ácidos nucleicos 5: e349. doi: 10.1038 / mtna.2016.58
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Engel, L. A., Jing, Z., O’Brien, D. E., Sun, M., and Kotzbauer, P. T. (2010). La función catalítica de PLA2G6 se ve afectada por mutaciones asociadas con distrofia neuroaxonal infantil, pero no por distonía-parkinsonismo. PLoS One 5: e12897. doi: 10.1371 / journal.ponga.0012897
PubMed Resumen | Texto Completo Cruzado
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (1993). en PLA2G6-Associated Neurodegeneration, eds M. P. Adam, H. H. Ardinger, y R. A. Pagon, Seattle, WA: GeneReviews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Afectación del SNC en la enfermedad de Fabry: estudios clínicos y de imágenes antes y después de 12 meses de terapia de reemplazo enzimático. J. Heredar. Metab. Dis. 27, 229–240. doi: 10.1023 / B:BOLI.0000028794.04349.91
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Rather, M. I., Bhat, V., Gopinath, S., et al. (2016). Análisis genético de PLA2G6 en 22 familias indias con distrofia neuroaxonal infantil, distrofia neuroaxonal de inicio tardío atípico y complejo de parkinsonismo distónico. PLoS One 11: e0155605. doi: 10.1371 / journal.ponga.0155605
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
LaFountaine, J. S., Fathe, K., y Smyth, H. D. (2015). Aplicaciones terapéuticas y de entrega de tecnologías de edición de genes ZFNs. TALENs, y CRISPR / Cas9. Int. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Análisis de la variación genética de codificación de proteínas en 60.706 seres humanos. Nature 536, 285-291. doi: 10.1038 / nature19057
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J.C., Ellinor, P. T., MacArthur, D. G., et al. (2017). La variación genética humana altera la especificidad de CRISPR-Cas9 con y sin objetivos en los loci implicados terapéuticamente. Proc. Natl. Acad. Sci. U. S. A. 14, E11257-E11266. doi: 10.1073 / pnas.1714640114
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Levi, S., y Finazzi, D. (2014). Neurodegeneración con acumulación de hierro en el cerebro: actualización de los mecanismos patógenos. Delantero. Pharmacol. 5:99. doi: 10.3389 / fphar.2014.00099
Texto completo de CrossRef / Google Scholar
Liou, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Localización mitocondrial de la ciclooxigenasa-2 y la fosfolipasa A2 independiente del calcio en células cancerosas humanas: implicación en la resistencia a la apoptosis. Exp. Celda Res. 306, 75-84. doi: 10.1016 / j. yexcr.2005.01.011
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Macauley, S.L., y Sands, M. S. (2009). Terapia de reemplazo enzimático dirigida al SNC prometedora para enfermedades de almacenamiento lisosomal. Exp. Neurol. 218, 5–8. doi: 10.1016 / j. expneurol.2009.03.040
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Mehnaaz, L. (2016). Neurodegeneración con acumulación de hierro cerebral (NBIA) anteriormente enfermedad de Hallervorden-Spatz. J. Assoc. Phys. India 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K., Morton, J. E. V., Gregory, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, que codifica una fosfolipasa A2, está mutada en trastornos neurodegenerativos con alto contenido de hierro cerebral. NAT. Genet. 38, 752–754. doi: 10.1038 / ng1826
Resumen de PubMed / Texto completo de CrossRef | Google Scholar
Murphy, M. P. (2009). Cómo las mitocondrias producen especies reactivas de oxígeno. Bioquímica. J. 417, 1-13. doi: 10.1042 / BJ20081386
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C., and Faria, A. (2016). Neurodegeneración con acumulación de hierro en el cerebro: reporte de un caso. Dement. Neuropsicol. 10, 160–164. doi: 10.1590 | S1980-5764-2016DN1002014
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Nishiyama, J., Mikuni, T., and Yasuda, R. (2017). Edición del genoma mediada por virus a través de reparación dirigida por homología en células mitóticas y postmitóticas en el cerebro de mamíferos. Neurona 96, 755.e5-768.e5. doi: 10.1016 / j. neurona.2017.10.004
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., and Farooqui, A. A. (2005). Distribución de la fosfolipasa A2 independiente del calcio (iPLA2) en el cerebro de mono. J. Neurocitol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Pardridge, W. M. (2002). Drogas y genes dirigidos al cerebro con caballos de Troya moleculares. NAT. Modif. De La Droga Descubierta. 1, 131–139. doi: 10.1038 / nrd725
Resumen de PubMed / Texto completo de CrossRef | Google Scholar
Pardridge, W. M. (2005a). Biología molecular de la barrera hematoencefálica. Mol. Biotechnol. 30, 57–70. doi: 10.1385 / MB:30:1:057
Texto completo de CrossRef / Google Scholar
Pardridge, W. M. (2005b). The blood-brain barrier: bottleneck in brain drug development (en inglés). NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Fosfolipasas A2 independientes del calcio y su papel en procesos biológicos y enfermedades. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Corrección de la enfermedad mediante terapia génica mediada por AAV en un nuevo modelo de mucopolisacaridosis tipo IIID en ratones. Zumbido. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg / ddx058
Resumen de PubMed / Texto completo de CrossRef | Google Scholar
Scott, D. A., y Zhang, F. (2017). Implications of human genetic variation in CRISPR-based therapeutic genome editing (en inglés). NAT. Mediterráneo. 23:1095. doi: 10.1038/nm.4377
Resumen de PubMed | Texto Completo de CrossRef | Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H., Song, K., and Ma, Z. A. (2006). La fosfolipasa A2 independiente del calcio localiza y protege las mitocondrias durante la inducción apoptótica por la estaurosporina. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Smith, A. J., Bainbridge, J. W., and Ali, R. R. (2012). Terapia de suplementación génica para formas recesivas de distrofias retinianas hereditarias. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Sorrentino, N. C., D’Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Una sulfamidasa altamente secretada diseñada para atravesar la barrera hematoencefálica corrige las lesiones cerebrales de ratones con mucopolisacaridosis tipo IIIA. EMBO Mol. Mediterráneo. 5, 675–690. doi: 10.1002 / mmem.201202083
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Sorrentino, N. C., y Fraldi, A. (2016). Objetivo cerebral en MPS-IIIA. Pediatra. Endocrinol. Apo. 13(Supl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., and Jones, S. S. (1997). Una nueva fosfolipasa A2 independiente del calcio citosólico contiene ocho motivos de anquirina. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Administración de Alimentos y Medicamentos de los Estados Unidos (2017). La FDA Aprueba el Primer Tratamiento para una Forma de Enfermedad de los Listones. Disponible en: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., y Gottlieb, R. A. (2002). La inhibición de la fosfolipasa A2 independiente del calcio mitocondrial (iPLA2) atenúa la pérdida de fosfolípidos mitocondriales y es cardioprotectora. Bioquímica. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
Resumen de PubMed / Texto completo de CrossRef | Google Scholar
Yu-Wai-Man, P. (2016). Manipulación genética para enfermedades neurodegenerativas hereditarias: ¿mito o realidad? Br. J. Ophthalmol. 100:1322. doi: 10.1136/bjophthalmol-2015-308329
PubMed Abstract | CrossRef Texto Completo | Google Scholar
Zhao, Z., Zhang, X., Zhao, C., Choi, J., Shi, J., Canción, K., et al. (2010). Protección de las células beta pancreáticas por grupo A TRAVÉS de la reparación de la peroxidación de la membrana mitocondrial mediada por la fosfolipasa A(2). Endocrinología 151, 3038-3048. doi: 10.1210/es.2010-0016
Resumen de PubMed / Texto completo de CrossRef / Google Scholar