Introdução
Infantil distrofia neuroaxonal (: INAD) é autossômica recessiva rara doença neurodegenerativa de frequência desconhecida. O início dos sintomas ocorre geralmente entre os 6 meses e os 3 anos de idade. Antes desse tempo, os lactentes desenvolvem-se normalmente. O primeiro sintoma do INAD pode ser a desaceleração da obtenção de marcos de desenvolvimento normais ou regressão em marcos de desenvolvimento (Ramanadham et al., 2015). Hipotonia tronco, estrabismo e nistagmo são sintomas precoces da doença (Gregory et al., 2017). A progressão da doença é rápida e, à medida que avança, perdem-se mais competências adquiridas. Os músculos logo se tornam hipotônicos e mais tarde tornam-se espásticos (Levi e Finazzi, 2014). Eventualmente, todo o controle muscular voluntário é perdido. A fraqueza muscular também pode levar a dificuldades na alimentação e respiração. Além do nistagmo, algumas crianças sofrem perda de visão. As funções cognitivas perdem-se gradualmente e a demência desenvolve-se. O tempo de vida é geralmente de 5 a 10 anos (Gregory et al., 1993; Jardim et al., 2004; Macauley and Sands, 2009).
Patologia Molecular
distrofia Neuraxonal Infantil pertence a uma família de doenças neurodegenerativas que inclui distrofia neuroaxonal atípica de início tardio (ANAD) e complexo de parkinsonismo da distonia (CPD). A maioria dos casos de INAD estão associados a mutações homozigóticas ou heterozigóticas compostas no gene PLA2G6 que afectam a actividade catalítica do seu produto proteico (Engel et al., 2010). O gene PLA2G6 codifica um grupo através da proteína A2 da fosfolipase independente do cálcio (PLA2G6 ou iPLA2ß, ∼85/88 kDa) com uma lipase e sete domínios contendo repetição da anquirina (Tang et al., 1997). O PLA2G6 hidrolisa a cadeia SN-2 acilo de fosfolípidos, gerando ácidos gordos livres e lisofosfolípidos.
os fosfolípidos na membrana interna das mitocôndrias são ricos em ácidos gordos insaturados na posição sn-2, particularmente cardiolipina (Seleznev et al., 2006). Estes ácidos gordos insaturados são particularmente vulneráveis às abundantes espécies reactivas de oxigénio produzidas pelas mitocôndrias (Murphy, 2009), resultando em fosfolípidos peroxidados na membrana interna da mitocôndria. PLA2G6 localiza-se na mitocôndria (Williams and Gottlieb, 2002; Liou et al., 2005) consistente com uma maior procura de hidrólise dos ácidos gordos peroxidados na posição sn-2 dos fosfolípidos, levando à remodelação dos fosfolípidos (Balsinde et al., 1995; Zhao et al., 2010). Quando o PLA2G6 é defeituoso, a integridade interna da membrana mitocôndria é danificada. PLA2G6 também localiza-se no axon (Ong et al., 2005; Seleznev et al., 2006) indicando uma maior procura localizada de remodelação fosfolípida também lá. A manifestação de tal acumulação no cérebro é única para áreas-chave do cérebro, como os gânglios basais que resultaram em vários nomes para a mesma patogênese molecular subjacente envolvendo PLAG26 (Mehnaaz, 2016; Nassif et al., 2016).
a análise de ultra-estrutura de neurónios em ratos nocaute de PLA2G6 é consistente com esta patologia molecular. Mitocôndrias com cristas ramificadas e tubulares, mitocôndrias com cristas degeneradas, axônios com colapso do citoesqueleto e perda parcial da membrana em terminais axon foram observados (Beck et al., 2011). A um nível microscópico, estas características aparecem como inchaços axonais e corpos esferóides em terminais pré-sinápticos (Figura 1) no sistema nervoso central ou periférico.
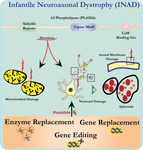
Figura 1. A distrofia neuroaxonal infantil (INAD) é uma doença neurodegenerativa relacionada com mutações no gene PLA2G6. Várias mutações de PLA2G6 levam a fosfolipase A2 disfuncional que leva a defeitos de membrana mitocondrial e axonal. Estes defeitos causam danos neuronais visualizados como tumefacção axonal e acumulação de esferóides pré-sinápticos. A substituição enzimática para restaurar funções, a substituição de genes ou a edição para corrigir o defeito do PLA2G6 são estratégias terapêuticas propostas.
Diagnóstico Molecular e Doenças Raras Paciente Empoderamento
Além das específicas clínicos, eletrofisiológicos e de recursos de imagens, antes a disponibilidade da próxima geração de sequenciamento, biópsias de pele mostrando axonal inchaços e esfera corpos em pré-sináptica terminais de central ou sistema nervoso periférico foram os critérios de diagnóstico para a confirmação de: INAD (Gregory et al., 2017; Iodice et al., 2017). Muitas vezes, várias biópsias eram necessárias para confirmar o diagnóstico. As famílias geralmente esperaram muitos anos por um diagnóstico. Com a diminuição do custo da sequenciação de genes e genomas, a disponibilidade de testes de painel de genes específicos com laboratórios de diagnóstico para doenças neurológicas não diagnosticadas, e a crescente conscientização dos médicos sobre a disponibilidade de diagnósticos genéticos, as famílias estão recebendo o diagnóstico mais rapidamente; às vezes, dentro de um ano após o primeiro sintoma aparecer. As crianças dessas famílias ainda são jovens e as famílias são motivadas a fazer parcerias com cientistas para encontrar um tratamento para a doença de seus filhos. Para financiar o processo de pesquisa, um grupo de pais de pacientes INAD formou a Fundação INADcure. A fundação angariou fundos substanciais para a investigação e está em parceria com o Rare Genomics Institute para os orientar na concessão de bolsas de investigação. Curiosamente, uma análise genética de 2016 de 22 famílias indianas com INAD, ANAD e CPD descobriu que 10/22 famílias (45,45%) não tinham mutações na região de codificação genética PLA2G6 (Kapoor et al., 2016). A incapacidade de identificar mutações deletérias na região de codificação de PLA2G6 sublinha que os futuros esforços de diagnóstico molecular exigiriam sequenciamento genético completo para identificar mutações nas regiões intrónicas e regulamentares do gene PLA2G6 em doentes afetados pela INAD. Além disso, nos casos CAD em que não se observam mutações associadas ao PLA2G6, sugere-se que a causa da doença pode ser devida a mutações em genes que não o PLA2G, o que tem de ser explorado.
possíveis terapêuticas
a maioria das terapêuticas consideradas para uma doença rara como a INAD que envolve uma enzima defeituosa são a substituição enzimática, a substituição genética ou a correcção genética. Quando uma deficiência enzimática é causada por um defeito genético recessivo, assume-se que a substituição enzimática ou suplementação pode corrigir o problema (Smith et al., 2012; Yu-Wai-Man, 2016). No entanto, experiências para provar que terapias que fornecem o gene ou enzima correto irão resgatar o fenótipo INAD ainda estão para ser realizadas e testadas.
terapêutica de substituição enzimática (ERT)
uma vez que o cérebro é o principal órgão afectado no INAD, a terapêutica de substituição enzimática do INAD necessitaria muito provavelmente de perfusão da enzima no cérebro. Em 2017, Biomarin recebeu a aprovação do FDA para tripeptidyl peptidase 1 (cerliponase alfa) como um tratamento para a causa subjacente de Sarrafo de doença, deficiência de tripeptidyl peptidase (TPP1) um lysosomal enzima (U.S. Food Drug Administration, 2017). Tripeptidil peptidase 1 (cerliponase alfa; 59 kDa) é o primeiro ERT a ser administrado directamente no líquido cefalorraquidiano (LCR) do cérebro. Outros medicamentos ERT que podem ser administrados no LCR estão em ensaios clínicos (Jardim et al., 2004; Macauley and Sands, 2009).
a eficácia da tripeptidil peptidase 1 (cerliponase alfa) na capacidade de andar dos doentes com doença de Batten demonstra que a terapêutica de substituição enzimática para doenças que afectam o cérebro é teoricamente possível. Direccionar a enzima de substituição para a mitocôndria para o INAD será mais desafiador do que o alvo lisossómico que já tinha sido bem sucedido pelo IV ERT por muito tempo, como no caso da doença de Gaucher. Assim, ainda existem muitas questões biológicas específicas do INAD que precisam ser resolvidas:
* a terapia para mecanismos de necessidade do INAD que podem possivelmente transportar o PLA2G6 para a célula?
* se a enzima PLA2G6 não é 100% pura, quais são os seus contaminantes e são seguros?Haverá alguma complicação intravascular generalizada devido à perfusão enzimática de PLA2G6 no SNC? A dose de perfusão tem alguma relevância para estas complicações?
uma questão adicional é que a terapêutica de substituição enzimática requer a colocação inicial de um cateter intracerebroventricular e infusões frequentes. A colocação do cateter requer anestesia e as perfusões podem exigir anestesia, dependendo da cooperação dos pacientes. Dar resposta às preocupações dos neurologistas pediátricos e dos pais de crianças com INAD no que diz respeito à utilização de anestesia em doentes com INAD com informação adequada exigirá mais esforços.
terapia genética / substituição de genes
o gene humano PLA2G é de cerca de 70 kb com 17 exões e 2 exons alternativos. A transcrição mais longa de codificação de proteínas, no entanto, é de apenas 3,3 kb e a sequência de codificação de proteínas é pouco mais de 2,4 kb, que pode ser facilmente empacotada em uma carga viral vectorial. Vamos primeiro explorar outro distúrbio de armazenamento lisossômico no SNC que tem dados pré-clínicos para ser comparado com o INAD. A falta de dados pré-clínicos de Tre no INAD é uma lacuna de conhecimento significativa no campo, e é por isso que é crucial que observemos outros modelos pré-clínicos de doenças do SNC que estão a ser submetidos a estudos de terapia genética. Anteriormente, um estudo pré-clínico em ratinhos abordou a terapia genética para a doença de armazenamento lisossómico mucopolissacaridoses tipo IIIA (MPS-IIIA) embalando a versão correcta do gene da sulfamidase dentro de um vector viral (Sorrentino et al., 2013). O estudo aproveitou a crescente compreensão da barreira hematoencefálica (BBB), que regula ativamente o transporte de grandes moléculas do sangue para o SNC por um processo chamado transcitose (Pardridge, 2005b; Sorrentino e Frataldi, 2016).
Transcitose envolve endocitose de ligantes no lado luminal, mediada por receptores específicos do ligando (por exemplo, receptor de insulina, receptor de transferrina, receptor de lipoproteína de baixa densidade, etc.,) enriched on the capillary endothelium (Pardridge, 2005a). Isto é seguido pelo movimento da carga endocitosada através do citoplasma de endotélio e, finalmente, exocitose no lado abluminal (cérebro), entregando assim eficazmente a carga através do BBB (Pardridge, 2002). O estudo pré-clínico MPS-IIIA fabricou uma proteína da sulfamidase quimérica que continha um domínio de ligação BBB da apolipoproteína B para facilitar a absorção pelo endotélio e também um péptido sinal da iduronato-2-sulfatase para ajudar a uma exocitose eficiente para o lado abluminal do BBB (Sorrentino et al., 2013). Uma carga viral vectorial que codificava uma sulfamidase quimérica foi então carregada sobre um serótipo 2/8 do vírus adeno associado (AAV) que visava o fígado (Sorrentino e Frataldi, 2016). Assim, o fígado serviu como uma fábrica interna que fornecia um fornecimento constante de quimeric-sulfamidase, o que resultou num aumento de 10-15% na actividade da sulfamidase cerebral mesmo 7 meses após a terapia genética hepática. Este aumento dos níveis de actividade da sulfamidase cerebral levou a uma melhoria quantificável da patologia cerebral e dos resultados comportamentais no modelo do rato MPS-IIIA (Sorrentino et al., 2013).
se for realizado um estudo semelhante para o INAD, este responderia a várias questões críticas sobre a substituição enzimática para a terapêutica com INAD. Em alternativa, a administração intra-vascular ou intra-CSF de produtos de terapia genética à base de AAV9 pode visar directamente o SNC (Bey et al., 2017; Roca et al., 2017). No entanto, as múltiplas mutações que os pacientes INAD têm em seu gene PLAG26 fornece desafios únicos para a terapia genética. Embora o tamanho do gene PLA2G6 de 2-3 kb não deva colocar um problema para sua inserção em um vetor viral, no entanto, as complicações regulatórias da correção do PLA2G6 são difíceis de prever. As complicações regulamentares tornam-se incontroláveis, especialmente se a terapia genética não pode ser localizada adequadamente dentro dos tecidos-alvo. Terapia de substituição enzimática também pode ser um problema se alguns produtos mutantes acabam por ser negativo dominante para o tipo selvagem PLAG26.
a maioria dos desafios das terapias possíveis para o INAD provém do facto de se tratar de uma doença ultra-rara. No entanto, o facto de o INAD ter uma etiologia genética conhecida proporciona pistas para possíveis terapias. Além disso, qualquer terapia bem sucedida para o INAD receberá o estatuto de medicamento órfão e toda a protecção que recebe porque o INAD é uma doença rara. Assim, apesar de todos os desafios acima mencionados, o estatuto de medicamento órfão constitui um forte incentivo para os investigadores em doenças raras e para a indústria da biotecnologia, para não falar de quando a causa genética já é conhecida.
edição de genes/bases
a partir de 2017, foram observadas pelo menos 277 mutações missense no gene humano PLA2G6 (Lek et al., 2016). Apenas uma pequena proporção de mutações PLA2G6 incluem mudanças de enquadramento, indels, mutações sem sentido e mutações nos locais de produção (Morgan et al., 2006). Assim, corrigir o gene na população de células alvo ou corrigir as bases de ADN mutadas é uma via terapêutica atraente. Ao longo dos anos, várias ferramentas foram desenvolvidas para a edição de genes, e a edição de genoma baseada em CRISPR/Cas9 está sendo saudada como uma tecnologia de ponta com um ensaio clínico planejado em 2018. Estas tecnologias utilizam uma proteína de ligação ao ADN que também pode clivear a cadeia de uma forma especificada para criar espaço para a inserção de uma nova sequência de ADN ou correcção da base deletéria de ADN (LaFountaine et al., 2015). A tecnologia CRISPR / Cas9 já produziu resultados terapêuticos surpreendentes em vários modelos pré-clínicos de doenças (por exemplo, distrofia muscular de Duchene, doenças metabólicas hepáticas, etc.,) (Dai et al., 2016). Além disso, uma crescente compreensão da variação humana está apresentando novos desafios à terapia genética e impulsionando a inovação para uma verdadeira personalização da edição de genes (Lessard et al., 2017; Scott and Zhang, 2017). Novas tecnologias, como vSLENDR, um vírus AAV, e Tecnologia mediada CRISPR/Cas9 para substituir genes defeituosos em neurônios e outras células do sistema nervoso estão empurrando tecnologias de edição de genes para novas fronteiras (Nishiyama et al., 2017).
conclusão
distrofia Infantil Neuroaxonal é uma doença neurodegenerativa grave com uma certa morbilidade e mortalidade. Esta doença rara oferece uma oportunidade emocionante para revalidar os modos disponíveis de terapia da próxima geração e gerar novos. Espera-se que a congruência emergente sobre os critérios de diagnóstico clínico para o INAD proporcione um impulso para o diagnóstico molecular melhorado. Espera-se que este progresso nos leve a desenvolver terapias a preços acessíveis que proporcionem uma melhoria quantificável na qualidade de vida dos doentes INAD e retardem ou melhorem a progressão da doença. Destacamos algumas histórias de sucesso na doença de Batten e as mucopolissacaridoses que nos fornecem a inspiração para fazer as perguntas certas para tornar a terapia INAD uma realidade. Além disso, o desenvolvimento de abordagens virais e não virais para a edição de genes com base em CRISPR/Cas9 deve também abrir novas vias terapêuticas para o INAD.
contribuições dos autores
PB e anteprojecto preparado pela FA. SR e AC editaram e adicionaram mais seções ao manuscrito. DF, AP e LP editaram o manuscrito.
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
Balsinde, J., Bianco, I. D., Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. A. (1995). A inibição da fosfolipase A2 independente do cálcio previne a incorporação de ácido araquidónico e a remodelação do fosfolípido nos macrófagos P388D1. Procedimento. Natl. Acad. Ciência. U. S. A. 92, 8527-8531. doi: 10.1073 / pnas.92.18.8527
PubMed Abstract | CrossRef Full Text | Google Scholar
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Alvos eficientes do SNC em ratinhos adultos por perfusão intratecal de AAV9-GFP de cadeia simples para terapia genética de distúrbios neurológicos. Gene Ther. 24:325. doi: 10.1038 / gt.2017.18
PubMed Abstract / CrossRef Full Text | Google Scholar
Dai, W.-J., Zhu, L.-Y., Yan, Z.-Y., Xu, Y., Wang, Q.-L., Lu, X.-J., et al. (2016). CRISPR-Cas9 para terapia genética in vivo: promessa e obstáculos. Mol. Ther. Ácidos nucleicos 5: e349. doi: 10.1038 / mtna.2016.58
PubMed Abstract / CrossRef Full Text | Google Scholar
Engel, L. A., Jing, Z., O’Brien, D. E., Sun, M., and Kotzbauer, P. T. (2010). A função catalítica do PLA2G6 é prejudicada por mutações associadas a distrofia neuroaxonal infantil, mas não por distonia-parkinsonismo. PLoS um 5: e12897. doi: 10.1371 / journal.pone.0012897
PubMed Abstract / CrossRef texto completo
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (1993). in PLA2G6-Associated Neurodegeneration, eds M. P. Adam, H. Ardinger, and R. A. Pagon, Seattle, WA: Generevews.
Google Scholar
Gregory, A., Kurian, M. A., Maher, E. R., Hogarth, P., and Hayflick, S. J. (2017). PLA2G6-Associated Neurodegeneration. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1675/
Google Scholar
Iodice, A., Spagnoli, C., Salerno, G. G., Frattini, D., Bertani, G., Bergonzini, P., et al. (2017). Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: an update for the diagnosis. Brain Dev. 39, 93–100. doi: 10.1016/j.braindev.2016.08.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Jardim, L., Vedolin, L., Schwartz, I. V., Burin, M. G., Cecchin, C., Kalakun, L., et al. (2004). Envolvimento do SNC na doença de Fabry: estudos clínicos e imagiológicos antes e após 12 meses de terapêutica de substituição enzimática. A J. Herdar. Metab. S. 27, 229–240. doi: 10.1023 / B: BOLI.0000028794.04349.91
PubMed Abstract | CrossRef Full Text | Google Scholar
Kapoor, S., Shah, M. H., Singh, N., Rather, M. I., Bhat, V., Gopinath, S., et al. (2016). Análise genética de PLA2G6 em 22 famílias indianas com distrofia neuroaxonal infantil, distrofia neuroaxonal atípica de início tardio e complexo de parkinsonismo distonia. PLoS One 11: e0155605. doi: 10.1371 / journal.pone.0155605
PubMed Abstract | CrossRef Full Text | Google Scholar
LaFountaine, J. S., Fathe, K., and Smyth, H. D. (2015). Entrega e aplicações terapêuticas de tecnologias de edição de genes ZFNs. TALENs e CRISPR/Cas9. T. J. Pharm. 494, 180–194. doi: 10.1016 / j. ijpharm.2015.08.029
PubMed Abstract | CrossRef Full Text | Google Scholar
Lek, M., Karczewski, K., Minikel, E., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291. doi: 10.1038/nature19057
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J. C., Ellinor, P. T. MacArthur, D. G., et al. (2017). A variação genética humana altera a especificidade CRISPR-Cas9 de focagem em loci implicado em termos terapêuticos. Procedimento. Natl. Acad. Ciência. U. S. A. 14, E11257–E11266. doi: 10.1073 / pnas.1714640114
PubMed Abstract | CrossRef Full Text | Google Scholar
Levi, S., and Finazzi, D. (2014). Neurodegeneração com acumulação de ferro cerebral: actualização sobre os mecanismos patogénicos. Frente. Farmacol. 5:99. doi: 10.3389 / fphar.2014.00099
CrossRef Texto Completo | Google acadêmico
Liu, J. Y., Aleksic, N., Chen, S. F., Han, T. J., Shyue, S. K., Wu, K. K., et al. (2005). Localização mitocondrial da ciclooxigenase-2 e fosfolipase A2 independente do cálcio em células cancerígenas humanas: implicação na resistência à apoptose. Expo. Cell Res. 306, 75-84. D. N.: 10.1016 / j. yexcr.2005. 01. 011
PubMed Abstract | CrossRef Full Text | Google Scholar
Macauley, S. L., And Sands, M. S. (2009). Terapia de substituição enzimática dirigida ao SNC para doenças de armazenamento lisossómico. Expo. Neurol. 218, 5–8. doi: 10.1016 / j. expneurol.2009.03.040
PubMed Abstract | CrossRef Full Text | Google Scholar
Mehnaaz, L. (2016). Neurodegeneração com acumulação de ferro cerebral (NBIA), anteriormente doença de Hallervorden – Spatz. J. Assoc. Phys. India 64: 132.
Google Scholar
Morgan, N. V., Westaway, S. K., Morton, J. e. V., Gregory, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, codificando uma fosfolipase A2, sofre uma mutação em distúrbios neurodegenerativos com ferro cerebral elevado. Conversao. Genet. 38, 752–754. doi: 10.1038/ng1826
PubMed Abstract / CrossRef Full Text | Google Scholar
Murphy, M. P. (2009). Como as mitocôndrias produzem espécies reactivas de oxigénio. Bioquímica. J. 417, 1-13. doi: 10.1042 / BJ20081386
PubMed Abstract / CrossRef Full Text | Google Scholar
Nassif, D., Pereira, J. S., Spitz, M., Capitão, C., e Faria, A. (2016). Neurodegeneração com acumulação de ferro cerebral: a case report. Dement. Neuropsicol. 10, 160–164. doi: 10.1590 / S1980-5764-2016DN1002014
PubMed Abstract | CrossRef Full Text | Google Scholar
Nishiyama, J., Mikuni, T., and Yasuda, R. (2017). Edição do genoma mediada pelo vírus via reparação homológica em células mitóticas e pós-mitóticas no cérebro de mamíferos. Neuron 96, 755.e5-768.e5. doi: 10.1016 / j. neuron.2017.10.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Ong, W.-Y., Yeo, J.-F., Ling, S.-F., And Farooqui, A. A. (2005). Distribuição da fosfolipase A2 (iPLA2) independente do cálcio no cérebro do macaco. J. Neurocytol. 34, 447–458. doi: 10.1007 / s11068-006-8730-4
PubMed Abstract | CrossRef texto completo | Google Scholar
Pardridge, W. M. (2002). Droga e gene a apontar para o cérebro com cavalos de Tróia moleculares. Conversao. Rev. Drug Discov. 1, 131–139. doi: 10.1038/nrd725
PubMed Abstract | CrossRef Full Text | Google Scholar
Pardridge, W. M. (2005a). Biologia Molecular da barreira hemato-encefálica. Mol. Biotechnol. 30, 57–70. doi: 10.1385 / MB:30: 1:057
CrossRef texto integral | Google Scholar
Pardridge, W. M. (2005b). A barreira sangue-cérebro: gargalo no desenvolvimento de medicamentos cerebrais. NeuroRx 2, 3-14.
Google Scholar
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., Lei, X., et al. (2015). Fosfolipases A2 independentes do cálcio e seus papéis em processos biológicos e doenças. J. Lipid Res. 56, 1643-1668. doi: 10.1194 / jlr.R058701
PubMed Abstract | CrossRef Full Text | Google Scholar
Roca, C., Mota, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., et al. (2017). Correcção da doença por terapia genética mediada pela AAV num novo modelo de ratinho de mucopolissacaridose tipo IIID. Cantarolar. Mol. Genet. 26, 1535–1551. doi: 10.1093 / hmg / ddx058
PubMed Abstract / CrossRef Full Text | Google Scholar
Scott, D. A., and Zhang, F. (2017). Implications of human genetic variation in CRISPR-based therapeutic genome editing. Conversao. Med. 23:1095. doi: 10.1038 / nm.4377
PubMed Abstract / CrossRef Full Text | Google Scholar
Seleznev, K., Zhao, C., Zhang, X. H.( 2006). A fosfolipase A2 independente do cálcio localiza-se e protege a mitocôndria durante a indução apoptótica pela staurosporina. J. Biol. Chem. 281, 22275–22288. doi: 10.1074 / jbc.M604330200
PubMed Abstract | CrossRef Full Text | Google Scholar
Smith, A. J., Bainbridge, J. W., and Ali, R. R. (2012). Terapia de suplementação genética para formas recessivas de distrofias hereditárias da retina. Gene Ther. 19, 154–161. doi: 10.1038 / gt.2011.161
PubMed Abstract / CrossRef Full Text | Google Scholar
Sorrentino, N. C., D’Orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). Uma sulfamidase altamente secretada, concebida para atravessar a barreira hemato-encefálica, corrige as lesões cerebrais de ratinhos com mucopolissacaridoses tipo IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002/emmm.201202083
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorrentino, N. C., e Fraldi, a. (2016). Alvo cerebral na MPS-IIIA. Pediatra. Endocrinol. Rev. 13 (Suppl. 1), 630–638.
Google Scholar
Tang, J., Kriz, R. W., Wolfman, N., Shaffer, M., Seehra, J., and Jones, S. S. (1997). Uma fosfolipase A2 citosólica independente do cálcio contém oito motivos para a anquirina. J. Biol. Chem. 272, 8567–8575. doi: 10.1074 / jbc.272.13.8567
PubMed Abstract | CrossRef Full Text | Google Scholar
U. S. Food and Drug Administration (2017). A FDA aprova o primeiro tratamento para uma forma de doença de Batten. Disponível em: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm555613.htm
Williams, S. D., and Gottlieb, R. A. (2002). A inibição da fosfolipase A2 (iPLA2) independente do cálcio mitocondrial atenua a perda de fosfolipídeo mitocondrial e é cardioprotetora. Bioquímica. J. 362 (Pt 1), 23-32. doi: 10.1042 / bj3620023
PubMed Abstract | CrossRef Full Text | Google Scholar
Yu-Wai-Man, P. (2016). Manipulação genética para doenças neurodegenerativas hereditárias: mito ou realidade? Br. J. Ophthalmol. 100:1322. doi: 10.1136/bjophthalmol-2015-308329
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Zhao, Z. Zhang, X., Zhao, C., Choi, J., Shi, J., Música, K., et al. (2010). Protecção das células beta pancreáticas por grupo VIA reparação da peroxidação da membrana mitocondrial mediada pela fosfolipase A(2). Endocrinology 151, 3038-3048. doi: 10.1210 / pt.2010-0016
PubMed Abstract / CrossRef Full Text | Google Scholar