Quantitative Messungen physiologischer Merkmale wie Enzymaktivität werden häufig als Aktivitätseinheiten pro Milligramm Protein ausgedrückt. Obwohl zahlreiche Assays zur Messung des Proteingehalts entwickelt wurden, einschließlich der kolorimetrischen Assays von Amido Black (1), Biuret (2), Bicinchoninsäure (3) und Coomassie Blue (4,5), werden der Lowry-Assay (6) oder seine Modifikationen (7,8) häufiger verwendet als andere Assays (9). Der Lowry-Assay ist einfach, sensitiv und präzise und das am häufigsten zitierte (10) Verfahren zur quantitativen Proteinbestimmung.
Eine Vielzahl von Verbindungen, die mit Folin-Ciocalteu-Phenol (Folin’s) -Reagenz (11) reagieren, sind eine Quelle potenzieller Interferenzen in Lowry- und modifizierten Lowry-Protein-Assays. Glücklicherweise ist für die meisten Verbindungen (6,7) mit Ausnahme von Lipiden (12), Detergenzien (13) und Farbstoffen (14) eine Korrektur durch einen geeigneten Rohling ausreichend. Schwierigkeiten bei der Bestimmung von Proteinen in Gegenwart von Lipiden und Detergenzien (die bei der Solubilisierung von Fettgewebe, Myelin und Skelettmuskeln verwendet werden) wurden durch den modifizierten Lowry-Assay überwunden (15; in diesem Artikel als U-1988-Assay bezeichnet, 16). Farbstörungen bei der Bestimmung des Proteingehalts in Rotwein (14,17,18) wurden durch aufwendige Chromatographie überwunden. Der obige Ansatz ist umständlich und nicht sehr praktisch für die Handhabung einer großen Anzahl von Proben. Keiner der bekannten Proteinassays war geeignet, Proteine in gefärbten biologischen Proben z.B., gefärbtes Obst und Gemüse, Rotwein, pigmentierte Mikroben und Wiederkäuergalle.
Unsere Entwicklung des U-2012-Assays aus seinen Vorgängern U-1988 und Lowry hat drei wesentliche Vorteile erzielt: (i) Bequemlichkeit durch Stabilität der Reagenzformulierungen, (ii) Messung von Proteinen sowohl in farblosen als auch in gefärbten biologischen Proben ohne Beeinträchtigung der Empfindlichkeit und (iii) Analyse von Proteinen bei sehr niedrigen Konzentrationen. Dieser neuartige Assay wird zur quantitativen Bestimmung von Protein sowohl in farblosen als auch in gefärbten biologischen Probenhomogenaten anwendbar sein, einschließlich solcher, die reich an Lipiden (z. B. Avocado) sind und schwer zu homogenisieren sind.
- Materialien und Methoden
- Biologische Proben – Rote Beete, Heidelbeere und Rotwein
- Chemische Reagenzien
- Verbesserungen des U-1988-Assays
- Der U-2012-Assay
- Schätzung der Farbinterferenzen im U-2012-Assay
- Die Standardkurve und ihre Parameter
- Berechnung des Proteingehalts in den Homogenaten
- Ergebnisse und Diskussion
- Verbesserungen im U-1988-Assay
- Proteinabschätzung in gefärbten biologischen Proben
- Proteinextraktion
- Störende Substanzen eliminieren
- Standardkurven und ihre Parameter
- Proteingehalt des gefärbten Homogenats
- Danksagung
- Konkurrierende Interessen
- Ergänzende Daten
Materialien und Methoden
Biologische Proben – Rote Beete, Heidelbeere und Rotwein
Rote Beete und Heidelbeere Homogenate wurden hergestellt, wie im Ergänzungsmaterial beschrieben. Rotwein erforderte vor dem U-2012-Assay keine Proteinextraktion.
Chemische Reagenzien
Alle chemischen Reagenzien außer Natriumhypochlorit und Perchlorsäure (PCA) wurden von Sigma oder Sigma-Aldrich (St Louis, MO. USA). Natriumhypochlorit wurde von Acros Organic, New Jersey, USA, hergestellt. PCA wurde von BDH (England) bezogen.
Verbesserungen des U-1988-Assays
Der Wechsel von Carbonat- zu Phosphatpuffer bei pH 12,0 verbesserte die Reagenzstabilität und führte zu einer geringen Erhöhung der Empfindlichkeit. Acetonitril wurde eingeführt, um detergenzinduzierte Blasen zu vermeiden. NaOH ersetzte KOH, um eine Ausfällung im Proteinassay zu vermeiden. Darüber hinaus wurde die Effizienz durch die Kombination verschiedener Komponenten des Lowry-Reagenzes in einem Reagenzienmix gesteigert.
Der U-2012-Assay
Ausführliche Informationen zum U-2012-Assay finden Sie im ergänzenden Material. Das in Abbildung 1 kurz zusammengefasste Protokoll beschreibt die Verarbeitung von Rotwein und die Homogenate von Rote Beete und Blaubeere und enthält die Verbesserungen des U-1988-Assays. Der U-2012-Assay wurde für unverarbeitete, verarbeitete und Reverse-Processed-Proteine (H2O2-Behandlung gefolgt von TCA- oder PCA-Präzipitation) eingesetzt. Assays wurden an BSA, Carboanhydrase, Cytochrom C, Isocitratdehydrogenase, Lysozym und Trypsin zur Entwicklung von Standardkurven und in farbigen biologischen Proben durchgeführt. Die Bestimmung der Proteine in den biologischen Proben erfolgte durch Kalibrierung auf entsprechende Standardkurven.
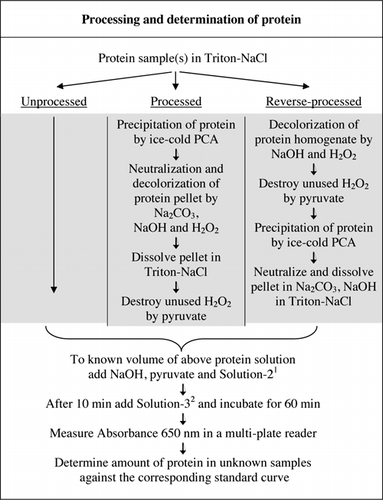
1Solution-2 enthielt Kupfersulfat (CuSO4.5H2O), Na-K-Tartrat, SDS und Acetonitril in 100 mM Phosphatpuffer (pH 12,0). 2Für Solution-3 wurde das Phenolreagenz von Folin-Ciocalteu verdünnt 1:1 mit entionisiertem Wasser kurz vor Gebrauch.
Schätzung der Farbinterferenzen im U-2012-Assay
Die Farbinterferenz wurde durch Vergleich der Absorptionsfähigkeit von verarbeiteten und unverarbeiteten Rote-Bete-, Blaubeer- und Rotweinproben mit und ohne Verwendung von Folin-Reagenz bestimmt, wie in Abbildung 1 beschrieben. Das Verhältnis wurde verwendet, um das Ausmaß der Interferenz festzustellen, wobei Abs1 die Absorption von unverarbeiteten Proben mit Folin-Reagenz ist; Abs2 ist die Absorption von unverarbeiteten Proben ohne Folin-Reagenz; Abs3 ist die Absorption von verarbeiteten Proben mit Folin-Reagenz; und Abs4 ist Absorption von verarbeiteten Proben ohne Folin-Reagenz.
Die Standardkurve und ihre Parameter
Lösung-1B und 1C, die im Abschnitt Rezepte des beigefügten Ergänzungsmaterials beschrieben sind, wurden für die Entwicklung der Standardkurven verwendet. Die Konzentration von BSA und die entsprechenden Extinktionswerte wurden mit einem X-Y-Streudiagramm aufgetragen. Die Form dieses Diagramms (Abbildung 2) zeigt eine Sättigungsreaktion bei höheren Konzentrationen mit einer sehr begrenzten anfänglichen linearen Reaktion. Dies war eine bevorzugte Kurvenform, die zuvor berichtet wurde (20). Anfangs wurde dies mit einer Exponentialform modelliert (19), aber spätere Studien zeigten, dass eine rechteckige Hyperbel eine verbesserte Ausrichtung auf die Reaktion ergab, insbesondere bei niedrigeren Konzentrationen. Diese letztere Form wurde nun standardisiert und die folgende Drei-Parameter-Gleichung wurde verwendet, um die Absorptions-Protein-Konzentrations-Beziehung zu beschreiben:

Konz = Proteinkonzentration,
A = Absorption bei Konz,
A0 = Absorption bei Nullkonzentration,
AM= Absorption bei maximaler Konzentration,
C50= Konzentration = Absorption
(AM + A0)/2.
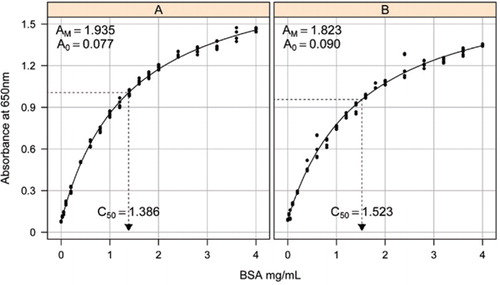
Die Schätzung der Standardkurve zwischen der Extinktion (650 nm) und den Konzentrationen von BSA-Protein wurde an drei technischen Replikaten zu (A) dem unverarbeiteten Proteinassay (Parameter A0 = 0,077, Am = 1,935, C50 = 1) durchgeführt.386) und (B) den prozessierten Proteinassay (Parameter A0=0,90, Am=1,823, C50=1,523). Ein rechteckiges Hyperbelmodell (Gleichung) wurde an die beobachteten Daten angepasst. Parameterschätzungen werden in den Kurven mit der C50-Schätzung auf der x-Achse (gepunktete schwarze Linie) identifiziert.
Parameter A0 wurde experimentell bestimmt, während AM und C50 mit der Toolbox-Add-In-Solver-Funktion von Microsoft Excel geschätzt wurden. Ein Versuchsparametersatz wurde verwendet, um die modellierte Extinktion bei jeder der Standardkonzentrationen (Conc) unter Verwendung der Gleichung zu berechnen . Solver wurde dann angewiesen, die verbleibende Standardabweichung zwischen der gemessenen und der modellierten Extinktion für den Standardsatz durch Anpassen von AM und C50 zu minimieren.
Wir haben beobachtet, dass die Beziehung zwischen Absorption und Konzentration über den gesamten Konzentrationsbereich eine nichtlineare Kurve aufweist, die wahrscheinlich auf eine Komponente der Lichtstreuung zurückzuführen ist, die mit zunehmender Proteinkonzentration bei der Messung der Absorption zunimmt. Eine schlechte lineare Anpassung bei geringer Absorption wurde auch von Coakley und James berichtet (20).
Berechnung des Proteingehalts in den Homogenaten
Assays wurden an verarbeitetem und unverarbeitetem Rotwein und Homogenaten von Rote Beete und Blaubeere durchgeführt. BSA- und andere Proteinproben wurden für geeignete Standardkurven identisch behandelt, um A0-, AM- und C50-Werte zu bestimmen. Diese Parameter wurden dann verwendet, um die Probenabsorption (A) in die Proteinkonzentration in jedem Homogenat umzuwandeln unter Verwendung von:

Da die Gleichung eine sättigende Form hat, verringert sich die Empfindlichkeit, wenn die Absorption (A) und damit die Konzentration zunimmt. Fehler in Proteinschätzungen können minimiert werden, indem die Konzentrationen von Homogenaten im Assay so eingestellt werden, dass sie den C50-Wert nicht übermäßig überschreiten.
Der Homogenat-Konz-Wert wurde dann unter Verwendung der folgenden Formel in die Gewebeproteinkonzentration (Gewebekonz in mg/g Gewebe) umgerechnet:

wo Homogenatkonz (in mg Protein/ml) für jede Vorkonzentration oder Verdünnung während des Tests korrigiert wurde. Der Homogenatanteil betrug 100 g Gewebe, homogenisiert auf ein Gesamtvolumen von 200 ml (in unserem Fall 50%).
In einer separaten Studie wurde ein rechteckiges Hyperbelmodell unter Verwendung des nichtlinearen Mischeffektpakets (NLME) (21) in R (22) angepasst (Abbildung 2). Jede BSA-Lösung, die unabhängig im Labor hergestellt wurde, wurde als Zufallseffekt mit einem gemeinsamen A0, aber unterschiedlichen AM- und C50-Koeffizienten modelliert. Dies modelliert die Hierarchie von biologischen Probenreplikaten und technischen Assayreplikaten.
Ergebnisse und Diskussion
Verbesserungen im U-1988-Assay
Die Einschränkung des U-1988- und des Lowry-Assays ist die Instabilität des Reagenzes auf Carbonatbasis. Der Carbonatpuffer (pH 11,4 bei 2% = 188,7 mM) in U-1988 wurde durch 40 mM Phosphat bei pH-Werten im Bereich von 11,4 bis 12,5 ersetzt. Anfängliche Steigungen aus den Standardkurven des Protein-Assays unter Verwendung von BSA bei 0,5 mg BSA / ml und 1,0 mg BSA / ml wurden berechnet. Die anfänglichen Steigungen mit Phosphatpuffern bei pH 11,4 und bei seinem optimalen pH 12,0 waren 99 x10-6 und 197 x10-6. Die Steigung für den Carbonatpuffer (pH 11,4) betrug 162 x10-6.Da der Steigungswert ein direkter Hinweis auf die Assay-Empfindlichkeit ist, Phosphatpuffer (pH 12.0) wurde gewählt, um Carbonatpuffer zu ersetzen, was eine 25% ige Erhöhung der Empfindlichkeit ergab.
Eine größere Stabilität wurde durch Erhöhung der Konzentration des Phosphatpuffers auf 100 mM erreicht. Die resultierende Phosphat/CuSO4/Na-K-tartrat-Lösung war bei Raumtemperatur zwei Wochen lang stabil, deutlich länger als die Carbonat /CuSO4/Na-K-tartrat-Lösung, die täglich vor dem Proteinassay hergestellt werden muss. Für alle zukünftigen Versuche wurde 100 mM Phosphat (pH 12,0) zur Herstellung der CuSO4/Na-K-Tartrat-Lösung verwendet. Wir glauben, dass der Ersatz von Carbonat durch Phosphat den Komfort des U-2012-Assays verbessern wird.
Detergenzinduzierte Blasen werden zu einer Hauptfehlerquelle bei Absorptionsmessungen bei Verwendung eines Multi-Well-Plattenlesegeräts (kein Problem bei Küvetten). Diese Blasen wurden durch Zugabe einer Reihe von polaren Lösungsmitteln (z.B. Aceton, Acetonitril, Ethanol und Methanol) erheblich reduziert. Acetonitril, das polarste dieser Lösungsmittel (23), wurde aufgrund seiner Wirksamkeit ausgewählt und in Solution-2 aufgenommen (siehe Abbildung 1 Beschriftung und Rezepte Abschnitt des ergänzenden Materials).
Phosphatpuffer, CuSO4, Na-K-Tartrat, SDS und Acetonitril können einzeln zugegeben werden und die Reihenfolge ihrer Zugabe beeinflusst die resultierende Absorption nicht. Die Verwendung einer vorgemischten Lösung erhöht jedoch den Komfort weiter, insbesondere wenn eine große Anzahl von Proben untersucht werden soll. Wir gruppierten daher diese Komponenten der Assay-Mischung in Lösung-2 (Abbildung 1). Eine solche vorgemischte Lösung war für den ursprünglichen Lowry-Assay (6) aufgrund der Instabilität der Carbonatlösung nicht durchführbar. Der Versuch, Solution-3 in Solution-2 aufzunehmen, führte zu einer dramatischen Verringerung der Entwicklung der blauen Farbe und wurde nicht weiter betrachtet.
Proteinabschätzung in gefärbten biologischen Proben
Proteinextraktion
Proteine aus Rote Bete und Blaubeere wurden in Triton X-100-NaCl-Lösung mit milder Homogenisierung extrahiert. Solche Homogenate behalten ihre Enzymaktivitäten (15). Diese Extraktion war für Rotwein nicht erforderlich.
Störende Substanzen eliminieren
Bei farbigen Proben ist es notwendig, die Interferenz aufgrund der inhärenten Probenfarbe und anderer Nicht-Protein-Substanzen, die mit den Proteinreagenzien reagieren, vor dem kolorimetrischen Proteinassay zu entfernen. Die Neuheit von U-2012 besteht in der Entwicklung eines Entfärbungsprotokolls, das mit einem kolorimetrischen Proteinassay kompatibel ist.
Entfärbung von Farbpigmenten durch Natriumhypochlorit oder H2O2 und selektive Ausfällung von Proteinen durch PCA oder TCA wurden zur Entfernung von Störsubstanzen in Betracht gezogen. Natriumhypochlorit, H2O2, TCA und PCA wurden auf ihre Kompatibilität mit dem U-2012-Assay unter Verwendung von BSA als Testprotein untersucht. Zwischen Natriumhypochlorit und H2O2 war nur H2O2 verträglich, da in Gegenwart von Hypochlorit ein Niederschlag gebildet wurde. Durch TCA oder PCA ausgefällte Proteine können nach ausreichender Neutralisation der Restsäure im Pellet mit U-2012 untersucht werden. Die Überlegenheit von PCA gegenüber TCA bei der Proteinfällung wurde berichtet (24,25). Im Gegensatz dazu in unserer vergleichenden Auswertung ergab ähnliche C50-Werte für PCA (1.395) und TCA (1.400). Wir bevorzugten PCA, weil es als vorgefertigte Lösung (70% v / v) leicht erhältlich ist und daher leicht auf die erforderliche Stärke verdünnt werden kann. TCA ist ein hygroskopischer Feststoff, der aufgrund seines variablen Wassergehalts schwer genau zu wiegen ist.
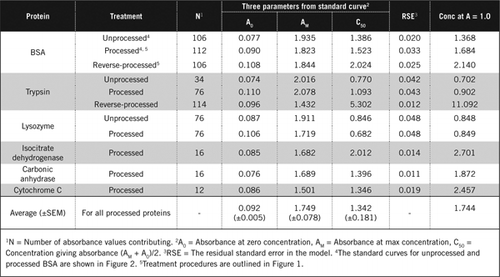
Es gibt zwei Möglichkeiten, PCA und H2O2 zu kombinieren. Für „verarbeitete“ Proteine folgte auf die PCA-Behandlung eine H2O2-Behandlung und für „Reverse-Processed“ -Proteine ging die H2O2-Behandlung der PCA-Präzipitation voraus. Vorteile der Verwendung von ‚prozessiertem‘ Protein waren die Entfernung einer Reihe von störenden Substanzen im Überstand und die mögliche Inaktivierung von proteolytischen Enzymen während der Probenvorbereitung. Dies wurde durch den Test von verarbeitetem und rückverarbeitetem Trypsin und BSA bestätigt (siehe Tabelle 1). Nur verarbeitete Proben wurden verwendet, um den tatsächlichen Proteingehalt farbiger biologischer Proben zu bestimmen
Sowohl PCA- als auch H2O2-Behandlungen farbiger Proben waren für die Interferenzeliminierung im U-2012-Assay erforderlich. Die saure Ausfällung allein der gefärbten Proben beseitigte die Interferenz nicht vollständig. Bei allen gefärbten Proben wurde etwas Farbe im Überstand verworfen, aber die Pellets waren auch gefärbt. Die Farbe wurde durch H2O2-Behandlung aus den Pellets entfernt. Alkalische Bedingungen waren sowohl für eine effektive Entfärbung durch H2O2 (26) als auch für die Farbentwicklung durch Folins Reagenz erforderlich, um sicherzustellen, dass die Proteinspiegel korrekt gemessen werden. Obwohl sowohl NaOH als auch KOH die erforderliche Alkalität liefern konnten, war nur NaOH mit dem U-2012-Assay kompatibel. In Gegenwart von KOH bildete sich ein Niederschlag. In Pellets wurde die PCA mit Na2CO3 und NaOH neutralisiert (27). Während des Assays wurde zusätzliche NaOH zugegeben; das optimierte Volumen lag zwischen 50 und 70 µL (60 µL wurden routinemäßig verwendet); siehe Abbildung 1.
Rote Beete, Heidelbeere und Rotwein wurden mit 15 µL 30% igem H2O2 unter 0,5 bzw. 2 h bei 50°C und Raumtemperatur entfärbt. Zwanzig Mikroliter 30% H2O2 für 1 h bei 50 ° C wurden verwendet, um stärker gefärbte Proben zu bewältigen. Die Oxidation von Substanzen wie Zuckern, die durch H2O2 bei 50 ° C an Proteine gebunden sind, scheint kritisch zu sein, da die Verarbeitung bei Raumtemperatur den Proteingehalt überschätzt. Im Fall von Rote Beete reduzierte die Verarbeitung bei 50 ° C die scheinbare Proteinschätzung auf 14% der unverarbeiteten, während die Verarbeitung bei Raumtemperatur diese Schätzung nur halbierte.
Aus den nach der Wasserstoffperoxidbehandlung durchgeführten kolorimetrischen Assays ging hervor, dass ein Teil von H2O2 bei der Entfärbung nicht genutzt wurde. In einer solchen Probe wurde die Endfarbe des Lowry-Assays teilweise zerstört. Es war daher notwendig, das verbleibende Wasserstoffperoxid vor dem Proteinassay zu zerstören. Es gibt zwei Quellen von H2O2 im U-2012-Assay; H2O2 zur Entfärbung hinzugefügt und H2O2 als Verunreinigung im Triton X-100 (0,22%, Produktinformation: Triton X-100, www.sigmaaldrich.com ). Wasserstoffperoxid wird üblicherweise durch das Enzym Katalase abgebaut. Der hohe pH-Wert des Protein-Assays würde jedoch bekannte Katalasen inaktivieren. Außerdem würde die Zugabe von Katalase zur Zugabe von zusätzlichem Protein führen. Wir entschieden uns für die chemische Zerstörung von H2O2 mit Pyruvat (28). Die Chemie der Pyruvat-H2O2-Wechselwirkungsgleichung ist gut etabliert (28,29). Pyruvat zerstört H2O2 bei Raumtemperatur gemäß der folgenden Reaktion:

Restliches H2O2 in der Pelletsuspension wurde durch Behandlung mit 0,9 M Pyruvat (1,5-fache H2O2-Konzentration) für 0,5 h bei Raumtemperatur zerstört. Um dem kontaminierenden H2O2 in Triton X-100 entgegenzuwirken, wurde im Proteinassay zusätzlich Pyruvat zugesetzt (Abbildung 1). Die Zugabe von Pyruvat ergab eine geringere Absorption für einen Nicht-Protein-Rohling . Wir schlagen vor, dass die Peroxidverunreinigung in Triton X-100 mit dem Acetonitril in Solution-2 reagiert, was zu einer etwas höheren Saugfähigkeit führt.
Die mit den gefärbten biologischen Proben verbundene Farbinterferenz kann nicht einfach durch Ausführen eines Proteintests in Abwesenheit des Folin-Reagenzes berücksichtigt werden. Die berechneten Verhältnisse (Abs1-Abs2) / (Abs3-Abs4) zeigten, dass die Störung durch die Probenfarbe für Rotwein (= 40) am höchsten und für Blaubeere ( = 6) und Rote Beete ( = 2) geringer war. Diese Interferenz führte zu den abnormal hohen Schätzungen der wahren Proteinspiegel; zum Beispiel die Proteinkonzentration unter Verwendung von unverarbeiteten und verarbeiteten Rote-Bete-Homogenaten (20,21 gegenüber 2,89 mg Protein / g Gewebe). Zusätzlich zu Farbstörungen enthalten Rotwein und Homogenate von Rote Beete und Blaubeere wahrscheinlich Substanzen, die mit Folins Reagenz im U-2012-Assay reagieren (e.g., kleine Peptide und komplexe Zucker). Diese wurden durch selektive Ausfällung von Proteinen mit eiskaltem PCA bei einer Endkonzentration von 5% entfernt (Abbildung 1).
Standardkurven und ihre Parameter
Standardkurven für unbearbeitete und verarbeitete BSA sind in Abbildung 2 dargestellt. Die abgeleiteten Parameter (A0, AM und C50) sind auch in Tabelle 1 für BSA und andere Proteine aufgeführt.
Die Ergebnisse zeigen, dass der verbleibende Standardfehler im Modell niedrig ist (0,012 bis 0,048), was auf die bessere Anpassung der Daten an den rechteckigen Hyperbeltrend hinweist. Zum Informationsvergleich zwischen verschiedenen Proteinen und deren Prozessierung wurden die Parameter auf die Konzentration für Extinktion = 1,0 bei 650 nm umgerechnet (rechte Spalte in Tabelle 1).
Diese Ergebnisse zeigen, dass der Proteinverlust (im Vergleich zu unverarbeitetem Protein) in verarbeiteten Proben geringer war als bei den umgekehrt verarbeiteten Proben. Dieser Verlust war im Fall von Trypsin deutlicher und kann aufgrund seiner autokatalytischen Aktivität während der Rückverarbeitung erklärt werden. Wir empfehlen, das ‚Processed‘-Protokoll (Supplementary Material) nur für biologische Proben zu befolgen, die wahrscheinlich proteolytische Enzyme enthalten.
Im ursprünglichen Lowry-Assay (6) und seiner modifizierten Version U-1988 (15) wurde nur der lineare Teil der Standardkurve, der durch Zeichnen der Extinktion gegen die Proteinmenge erhalten wurde, zur quantitativen Bestimmung des Proteins verwendet. Im U-2012-Assay verwenden wir die Daten effektiver, indem wir eine rechteckige Hyperbelgleichung anpassen, wie im Abschnitt Materialien und Methoden in Übereinstimmung mit Coakley und James (20) beschrieben.
Proteingehalt des gefärbten Homogenats
Die Proteinkonzentrationen in unbekannten Proben wurden durch Gleichung und gegen den verarbeiteten BSA-Standard und den Durchschnitt aller in Tabelle 1 aufgeführten verarbeiteten Proteine berechnet. Letzteres liegt näher an einer echten Schätzung für biologische Proben, die eine Mischung von Proteinen enthalten. Wir schätzten die Proteinmengen in Blaubeere und Rote Beete im Vergleich zu Rotwein auf das 60- bzw. 230-fache (Tabelle 2).
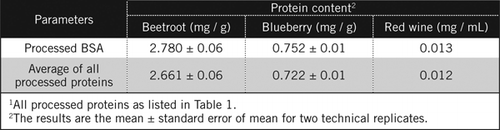
Wie BSA wurden Rotwein und 50% ige Homogenate von Rote Beete und Blaubeere durch PCA-Fällung und Entfärbung durch H2O2 verarbeitet (Abbildung 1). In diesem Stadium wurden die biologischen Proben 40 mal für Rotwein und 4 Mal für Rote Beete und Blaubeere konzentriert. In ähnlicher Weise wurde BSA (2 mg / ml) auch 4-mal auf 8 mg / ml konzentriert. Die Extinktion der gefärbten Proben, die nahe der Extinktion für C50 (für unverarbeitetes BSA) lag, wurde verwendet, um den Proteingehalt zu berechnen, wie durch Gleichung und beschrieben.
Zusammenfassend hat der U-2012-Assay stabile Reagenzien eingesetzt, eine verbesserte Empfindlichkeit (auch für farblose biologische Proben) bereitgestellt und farbinduzierte Interferenzen für farbige biologische Proben überwunden. Der U-2012-Assay ist nicht auf den linearen Teil der Reaktion zwischen Proteinkonzentration und Absorption beschränkt und nutzt Daten im nichtlinearen Bereich effizienter durch ein rechteckiges hyperbolisches Kurvenmodell, das mit einfachen Verfahren in Microsoft Excel an die Standards angepasst wird.
Danksagung
Autoren danken der Stiftung für Forschung, Wissenschaft und Technologie Neuseeland für finanzielle Unterstützung (C06X0809).
Konkurrierende Interessen
Die Autoren erklären keine konkurrierenden Interessen.
Ergänzende Daten
Die ergänzenden Daten zu diesem Artikel finden Sie auf der Website der Zeitschrift unter: www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R.S. und P.L. Pedersen. 1985. Bestimmung von Mikrogramm Protein in Gegenwart von Milligramm Lipid mit Amido black 10B. Anal. Biochem. 150:97–104.Crossref, Medline, CAS, Google Scholar
- 2. Gornall, A.G., C.J. Bardawill und M.M. David. 1949. Bestimmung von Serumproteinen mittels der Biuret-Reaktion. J. Biol. Chem. 177:751–766.Medline, CAS, Google Scholar
- 3. In diesem Fall ist es nicht möglich, die Ergebnisse zu überprüfen.. 1985. Messung von Protein mit Bicinchoninsäure. Anal. Biochem. 150:76–85.Crossref, Medline, CAS, Google Scholar
- 4. Bradford, M.M. 1976. Eine schnelle und empfindliche Methode zur Quantifizierung von Mikrogramm-Proteinmengen unter Verwendung des Prinzips der Protein-Farbstoff-Bindung. Anal. Biochem. 72:248–254.Crossref, Medline, CAS, Google Scholar
- 5. Zor, T. und Z. Selinger. 1996. Die Linearisierung des Bradford-Protein-Assays erhöht seine Sensitivität: theoretische und experimentelle Studien. Anal. Biochem. 236:302–308.Crossref, Medline, CAS, Google Scholar
- 6. Lowry, O.H., N.J. Rosbrough, A.L. Farr und R.J. Randall. 1951. Proteinmessung mit dem Folin Phenol Reagenz. In: J. Biol. Chem. 193:265–275.Medline, CAS, Google Scholar
- 7. Peterson, G.L. 1979. Überprüfung der Folinphenolproteinquantifizierungsmethode von Lowry, Rosebrough, Farr und Randall. Anal. Biochem. 100:201–220.Crossref, Medline, CAS, Google Scholar
- 8. Sapan, C.V., R.L. Lundablad und N.C. Price. 1999. Kolorimetrische Protein-Assay-Techniken. In: Biotechnol. Appl. Biochem. 29:99–108.Medline, CAS, Google Scholar
- 9. Okutucu, B., A. Dınçer, Ö. Habib, und F. Zıhnıoglu. 2007. Vergleich von fünf Methoden zur Bestimmung der Gesamtplasmaproteinkonzentration. In: J. Biochem. In: Biophys. Methoden 70: 709-711.Crossref, Medline, CAS, Google Scholar
- 10. Kresge, N., R.D. Simoni und R.L. Hill. 2005. Das am häufigsten zitierte Papier in der Verlagsgeschichte: Proteinbestimmung von Oliver H. Lowry. In: J. Biol. Chem. 25:280.Google Scholar
- 11. Everette, J.D., Q.M. Bryant, A.M. Green, Y.A. Abbey, G.W. Wangila und R.B. Walker. 2010. Gründliche Untersuchung der Reaktivität verschiedener Verbindungsklassen gegenüber dem Folin-Ciocalteu-Reagenz. J. Agric. Lebensmittel Chem. 58:8139–8144.Crossref, Medline, CAS, Google Scholar
- 12. Eichberg, J. und L.C. Mokrasch. 1969. Interferenz durch oxidierte Lipide bei der Proteinbestimmung nach dem Lowry-Verfahren. Anal. Biochem. 30:386–390.Crossref, Medline, CAS, Google Scholar
- 13. Dulley, J.R. und P.A. Trauern. 1975. Eine einfache Technik zur Beseitigung von Störungen durch Detergenzien in der Lowry-Methode der Proteinbestimmung. Anal. Biochem. 64:136–141.Crossref, Medline, CAS, Google Scholar
- 14. Brillouet, J.-M., M.-P. Belleville und M. Moutounet. 1991. Mögliche Protein-Polysaccharid-Komplexe in Rotweinen. Uhr. In: J. Enol. Wein. 42:150–152.CAS, Google Scholar
- 15. Upreti, G.C., R.A. Ratcliff und P.C. Riches. 1988. Proteinschätzung in Geweben mit hohem Lipidgehalt: Modifikationen der Lowry-Methode zur Proteinbestimmung. Anal. Biochem. 168:421–427.Crossref, Medline, CAS, Google Scholar
- 16. Upreti, G.C., C. Davis und J. Oliver. 1991. Herstellung repräsentativer Homogenate biologischer Gewebe: Wirkung von Salz auf die Proteinextraktion. Anal. Biochem. 198:298–301.Crossref, Medline, CAS, Google Scholar
- 17. Smith, M.R., M.H. Penner, S. E. Bennett und A.T. Bakalinsky. 2011. Quantitativer kolorimetrischer Test für Gesamtprotein auf den Rotwein Pinot Noir angewendet. J. Agric. Lebensmittel Chem. 59:6871–6876.Crossref, Medline, CAS, Google Scholar
- 18. Wigand, P., S. Tenzer, H. Schild, und H. Decker. 2009. Analyse der Proteinzusammensetzung von Rotwein im Vergleich zu Rosé- und Weißweinen durch Elektrophorese und Hochdruckflüssigkeitschromatographie-Massenspektrometrie (HPLC-MS). J. Agric. Lebensmittel Chem. 57:4328–4333.Crossref, Medline, CAS, Google Scholar
- 19. Upreti, G.C., Y. Wang, A. Sharrock, N. Feisst, M. Davy und B. Jordan. 2009. Ein stabiler und empfindlicher Proteinassay (U-2009 modifizierter Assay) für farbige biologische Proben. ComBiol., Neuseeland Endgültiges Programm Dezember 2009. Universität von Canterbury, Christchurch, Neuseeland.Google Scholar
- 20. Coakley, W.T. und C.J. James. 1978. Eine einfache lineare Transformation für die Folin-Lowry-Proteinkalibrierungskurve auf 1,0 mg / ml. Anal. Biochem. 85:90–97.Crossref, Medline, CAS, Google Scholar
- 21. Pinheiro, J.C. und D.M. Bates. 2000. Mixed-Effects-Modelle in S und S-PLUS, Statistik und Computing Series. In: Springer-Verlag, New York, NY.Google Scholar
- 22. R Entwicklung Kernteam. 2009. R: Eine Sprache und Umgebung für statistisches Rechnen. R Stiftung für Statistisches Rechnen, Wien, Österreich, ISBN 3-900051-07-0, URL http://www.R-project.org.Google
- 23. Khachik, F., G.R. Beecher, J.T. Vanderslice, und G. Furche. 1988. Flüssigchromatographische Artefakte und Peakverzerrung: Probe-Lösungsmittel-Wechselwirkung bei der Trennung von Carotinoiden. Anal. Chem. 60:807–811.Crossref, Medline, CAS, Google Scholar
- 24. Cernik, AA 1970. Bestimmung von Bleichelat mit Ethylendiamintetraessigsäure im Blut nach Ausfällung von Protein mit Perchlorsäure. Brite. In: J. Industry Med. 27:40–42.Medline, CAS, Google Scholar
- 25. Moughan, P.J., A.J. Darragh, W.C. Smith und C.A. Butts. 1990. Perchlorsäure und Trichloressigsäure als Fällungsmittel von Protein in endogenen Ileumverdauungen aus der Ratte. J. Sci. Lebensmittel Agric. 52:13–21.Crossref, CAS, Google Scholar
- 26. Galbács, Z.M. und L.J. Csányi. 1983. Alkali-induzierte Zersetzung von Wasserstoffperoxid. In: J. Chem. Soc., Dalton Trans. 11:2353–2357.Crossref, Google Scholar
- 27. Scopes, R.K. 1988. Proteinreinigung: Prinzipien und Praxis, zweite Auflage. Springer-Verlag New York Inc., New York, NY.Google Scholar
- 28. Upreti, G.C., K. Jensen, R. Munday, D.M. Duganzich, R. Vishwanath und J.F. Smith. 1998. Studien zur aromatischen Aminosäureoxidaseaktivität in RAM-Spermatozoen: Rolle von Pyruvat als Antioxidans. Anim. Reprod. Sci. 51:275–287.Crossref, Medline, CAS, Google Scholar
- 29. Holleman, M.A.F. 1904. Notice sur l’action de l’eau oxygenee sur les acids α-cetoniques et sur les dicetones 1.2. Recl. Trav. Chim. Pays-Bas Belg. 23:169–172.Querverweis, CAS, Google Scholar