Les mesures quantitatives des traits physiologiques tels que l’activité enzymatique sont souvent exprimées en unités d’activité par milligramme de protéine. Bien que de nombreux dosages aient été développés pour mesurer la teneur en protéines, y compris les dosages colorimétriques du Noir d’Amido (1), du Biuret (2), de l’Acide Bicinchoninique (3) et du Bleu de Coomassie (4,5), le test de Lowry (6) ou ses modifications (7,8) sont plus couramment utilisés que les autres dosages (9). Le test Lowry est simple, sensible et précis, et est la procédure la plus citée (10) pour la détermination quantitative des protéines.
Une grande variété de composés qui réagissent avec le réactif Folin-Ciocalteu phénol (Folin’s) (11) sont une source d’interférence potentielle dans les dosages de protéines Lowry et de protéines Lowry modifiées. Heureusement, les corrections à travers une ébauche appropriée sont suffisantes pour la plupart des composés (6,7) à l’exception des lipides (12), des détergents (13) et des substances colorées (14). Les difficultés de dosage des protéines en présence de lipides et de détergents (utilisés dans la solubilisation du tissu adipeux, de la myéline et des muscles squelettiques) ont été surmontées par le test de Lowry modifié (15; désigné dans cet article comme le test U-1988, 16). L’interférence de la couleur dans la détermination de la teneur en protéines du vin rouge (14,17,18) a été surmontée en utilisant une chromatographie extensive. L’approche ci-dessus est lourde et peu pratique pour manipuler un grand nombre d’échantillons. Aucun des dosages de protéines connus ne convenait à la mesure de protéines dans des échantillons biologiques colorés, p.ex., fruits et légumes colorés, vin rouge, microbes pigmentés et bile de ruminants.
Notre développement du test U-2012 à partir de ses prédécesseurs, le test U-1988 et le test Lowry a obtenu trois avantages majeurs (i) la commodité grâce à la stabilité des formulations de réactifs, (ii) la mesure des protéines dans des échantillons biologiques incolores et colorés sans compromettre la sensibilité, et (iii) le dosage des protéines à de très faibles concentrations. Ce nouveau dosage sera applicable à la détermination quantitative des protéines dans les homogénats d’échantillons biologiques incolores et colorés, y compris ceux riches en lipides (par exemple, l’avocat) et ceux difficiles à homogénéiser.
- Matériaux et méthodes
- Échantillons biologiques – betterave, myrtille et vin rouge
- Réactifs chimiques
- Les améliorations apportées au test U-1988
- L’essai U-2012
- L’estimation des interférences de couleur dans le test U-2012
- La courbe standard et ses paramètres
- Calcul de la teneur en protéines dans les homogénats
- Résultats et discussion
- Améliorations de l’essai U-1988
- Estimation des protéines dans des échantillons biologiques colorés
- Extraction des protéines
- Éliminer les substances interférantes
- Courbes standard et leurs paramètres
- Teneur en protéines de l’homogénat coloré
- Remerciements
- Intérêts concurrents
- Données supplémentaires
Matériaux et méthodes
Échantillons biologiques – betterave, myrtille et vin rouge
Les homogénats de betterave et de myrtille ont été préparés comme décrit dans le Matériel supplémentaire. Le vin rouge ne nécessitait pas d’extraction de protéines avant l’essai U-2012.
Réactifs chimiques
Tous les réactifs chimiques, à l’exception de l’hypochlorite de sodium et de l’acide perchlorique (PCA), ont été obtenus à partir de Sigma ou Sigma-Aldrich (St Louis, MO. USA). L’hypochlorite de sodium provient d’Acros Organic, New Jersey, États-Unis. La PCA a été obtenue auprès de BDH (Angleterre).
Les améliorations apportées au test U-1988
Le passage du tampon carbonate au tampon phosphate à pH 12,0 a amélioré la stabilité du réactif et a donné une légère augmentation de la sensibilité. L’acétonitrile a été introduit pour éviter les bulles induites par le détergent. NaOH a remplacé KOH pour éviter la précipitation dans le test de protéines. De plus, l’efficacité a été améliorée en combinant divers composants du réactif Lowry en un seul mélange de réactifs.
L’essai U-2012
Les détails complets de l’essai U-2012 sont fournis dans des documents supplémentaires. Le protocole, brièvement résumé à la figure 1, décrit le traitement du vin rouge et les homogénats de betterave et de myrtille, et comprend les améliorations apportées à l’essai U-1988. L’essai U-2012 a été utilisé pour les protéines non transformées, transformées et inversées (traitement H2O2 suivi d’une précipitation de TCA ou de PCA). Des dosages ont été réalisés sur la BSA, l’anhydrase carbonique, le cytochrome C, l’isocitrate déshydrogénase, le lysozyme et la trypsine pour l’élaboration de courbes standard et dans des échantillons biologiques colorés. La détermination des protéines dans les échantillons biologiques a été effectuée par calibrage selon des courbes standard appropriées.
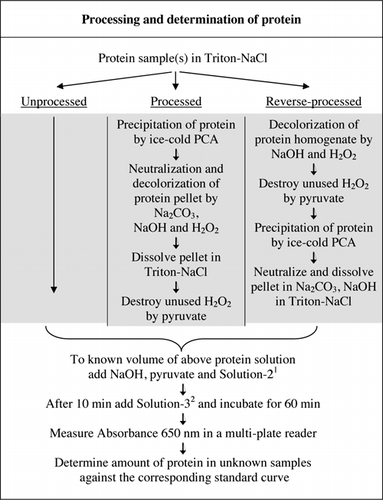
1Solution-2 contenait du sulfate de cuivre (CuSO4,5H2O), du Na-K-Tartrate, du SDS et de l’acétonitrile dans un tampon phosphate de 100 mM (pH 12,0). 2Pour la solution-3, le réactif phénol de Folin-Ciocalteu a été dilué 1:1 avec de l’eau désionisée juste avant utilisation.
L’estimation des interférences de couleur dans le test U-2012
Les interférences de couleur ont été déterminées en comparant l’absorption d’échantillons de betteraves, de myrtilles et de vin rouge transformés et non transformés avec et sans l’utilisation du réactif de Folin, comme décrit à la figure 1. Le rapport a été utilisé pour établir l’étendue de l’interférence, où Abs1 est l’absorbance des échantillons non traités avec le réactif de Folin; Abs2 est l’absorbance des échantillons non traités sans réactif de Folin; Abs3 est l’absorbance des échantillons traités avec le réactif de Folin; et Abs4 est l’absorbance des échantillons traités sans réactif de Folin.
La courbe standard et ses paramètres
Solution-1B et 1C décrits dans la section Recettes du Matériel supplémentaire ci-joint ont été utilisés pour l’élaboration des courbes standard. La concentration de BSA et les valeurs d’absorbance correspondantes ont été tracées à l’aide d’un graphe de dispersion X-Y. La forme de ce graphique (Figure 2) montre une réponse saturante à des concentrations plus élevées avec une réponse linéaire initiale très limitée. Il s’agissait d’une forme de courbe préférée signalée précédemment (20). Au départ, cela a été modélisé en utilisant une forme exponentielle (19), mais des études ultérieures ont montré qu’une hyperbole rectangulaire donnait un meilleur alignement avec la réponse, en particulier à des concentrations plus faibles. Cette dernière forme a maintenant été normalisée et l’équation à trois paramètres suivante a été utilisée pour décrire la relation absorbance-concentration en protéines:

Conc = Concentration en protéines,
A = Absorbance à Conc,
A0 = Absorbance à concentration nulle,
AM = Absorbance à concentration maximale,
C50 = Concentration donnant une absorbance
(AM + A0) / 2.
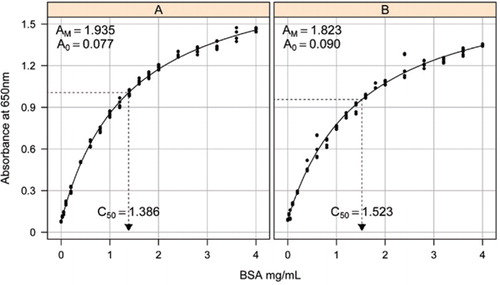
L’estimation de la courbe standard entre l’absorbance (650 nm) et les concentrations de protéine BSA a été faite sur trois répliques techniques à (A) l’essai de protéine non traitée (paramètres A0 = 0,077, Am = 1,935, C50 = 1.386), et (B) le dosage des protéines traitées (paramètres A0 = 0,90, Am = 1,823, C50 = 1,523). Un modèle d’hyperbole rectangulaire (équation) a été ajusté aux données observées. Les estimations des paramètres sont identifiées dans les courbes avec l’estimation C50 sur l’axe des abscisses (ligne noire pointillée).
Le paramètre A0 a été déterminé expérimentalement tandis que AM et C50 ont été estimés à l’aide de la fonction de solveur complémentaire Toolbox de Microsoft Excel. Un ensemble de paramètres d’essai a été utilisé pour calculer l’absorbance modélisée à chacune des concentrations standard (Conc) en utilisant l’équation. Le Solveur a ensuite été commandé pour minimiser l’écart-type résiduel entre l’absorbance mesurée et modélisée pour l’ensemble de normes en ajustant AM et C50.
Nous avons observé que la relation entre l’absorbance et la concentration présentait une courbe non linéaire sur toute la plage de concentration, ce qui est probablement dû à une composante de la diffusion de la lumière qui augmente à mesure que la concentration de protéines augmente lors de la mesure de l’absorbance. Un mauvais ajustement linéaire à faible absorbance a également été signalé par Coakley et James (20).
Calcul de la teneur en protéines dans les homogénats
Des dosages ont été effectués sur du vin rouge transformé et non transformé et des homogénats de betterave et de myrtille. Les échantillons de BSA et d’autres protéines ont été traités de manière identique pour des courbes standard appropriées afin de déterminer les valeurs A0, AM et C50. Ces paramètres ont ensuite été utilisés pour convertir l’absorbance de l’échantillon (A) en concentration de protéines dans chaque homogénéat en utilisant:

Comme l’équation a une forme saturante, la sensibilité diminue à mesure que l’absorbance (A), et donc la concentration, augmente. Les erreurs dans les estimations des protéines peuvent être minimisées en ajustant les concentrations d’homogénats dans le test afin qu’elles ne dépassent pas excessivement la valeur C50.
La valeur de l’homogénéat Conc a ensuite été convertie en concentration de protéines tissulaires (Conc tissulaire en mg/g de tissu) en utilisant la formule suivante:

lorsque l’homogénat Conc (en mg de protéine / mL) a été corrigé pour toute pré-concentration ou dilution au cours de l’essai. Le pourcentage d’homogénéité était de 100 g de tissu homogénéisé pour un volume total de 200 mL (dans notre cas 50%).
Dans une étude distincte, un modèle d’hyperbole rectangulaire a été ajusté en utilisant l’ensemble d’effets mixtes non linéaires (NLME) (21) dans R(22) (Figure 2). Chaque solution de BSA, fabriquée indépendamment en laboratoire, a été modélisée comme un effet aléatoire, avec un A0 commun mais des coefficients AM et C50 différents. Cela permet de modéliser la hiérarchie des répliques d’échantillons biologiques et des répliques d’essais techniques.
Résultats et discussion
Améliorations de l’essai U-1988
La limitation de l’essai U-1988 et de l’essai Lowry est l’instabilité du réactif à base de carbonate. Le tampon carbonate (pH 11,4 à 2% = 188,7 mM) dans U-1988 a été remplacé par du phosphate de 40 mM à des pH allant de 11,4 à 12,5. Les pentes initiales des courbes standard de l’essai protéique utilisant BSA à 0,5 mg BSA/mL et 1,0 mg BSA/mL ont été calculées. Les pentes initiales avec des tampons phosphatés à pH 11,4 et à son pH optimal 12,0 étaient respectivement de 99 x10-6 et 197 x10-6. La pente du tampon carbonate (pH 11,4) est de 162 x10-6.Étant donné que la valeur de pente est une indication directe de la sensibilité du test, le tampon phosphate (pH 12.0) a été choisi pour remplacer le tampon carbonate, donnant une augmentation de sensibilité de 25%.
Une plus grande stabilité a été obtenue en augmentant la concentration du tampon phosphate à 100 mM. La solution phosphate/CuSO4 / Na-K-tartrate résultante a été stable à température ambiante pendant deux semaines, considérablement plus longtemps que la solution carbonate / CuSO4 / Na-K-tartrate, qui doit être préparée quotidiennement avant le dosage des protéines. Pour toutes les expériences futures, 100 mm de phosphate (pH 12,0) ont été utilisés pour préparer la solution de CuSO4/Na-K-tartrate. Nous pensons que le remplacement du carbonate par du phosphate améliorera la commodité du test U-2012.
Les bulles induites par le détergent deviennent une source majeure d’erreur dans les mesures d’absorbance lors de l’utilisation d’un lecteur de plaques à puits multiples (pas un problème avec les cuvettes). Ces bulles ont été considérablement réduites par l’ajout d’un certain nombre de solvants polaires (par exemple, acétone, acétonitrile, éthanol et méthanol). L’acétonitrile, le plus polaire de ces solvants (23), a été choisi pour son efficacité et inclus dans la Solution-2 (voir la section Légende et Recettes de la Figure 1 du Matériel supplémentaire).
Le tampon phosphate, le CuSO4, le Na-K-tartrate, le SDS et l’acétonitrile peuvent être ajoutés individuellement et l’ordre de leur addition n’affecte pas l’absorbance résultante. Cependant, l’utilisation d’une solution prémélangée améliore encore la commodité, en particulier lorsque de grands nombres d’échantillons doivent être dosés. Nous avons donc regroupé ces composants du mélange de dosage dans la Solution 2 (Figure 1). Une telle solution prémélangée n’était pas réalisable pour l’essai initial de Lowry (6) en raison de l’instabilité de la solution de carbonate. La tentative d’inclure la Solution-3 dans la Solution-2 a entraîné une réduction spectaculaire du développement de la couleur bleue et n’a pas été envisagée plus avant.
Estimation des protéines dans des échantillons biologiques colorés
Extraction des protéines
Les protéines de betterave et de myrtille ont été extraites dans une solution de Triton X-100-NaCl avec une légère homogénéisation. De tels homogénéats conservent leurs activités enzymatiques (15). Cette extraction n’était pas nécessaire pour le vin rouge.
Éliminer les substances interférantes
Pour les échantillons colorés, il est nécessaire d’éliminer les interférences dues à la couleur inhérente de l’échantillon et aux autres substances non protéiques qui réagissent avec les réactifs protéiques avant le dosage colorimétrique des protéines. La nouveauté de U-2012 réside dans la conception d’un protocole de décoloration compatible avec un dosage colorimétrique des protéines.
La décoloration des pigments colorés par l’hypochlorite de sodium ou l’H2O2 et la précipitation sélective des protéines par le PCA ou le TCA ont été envisagées pour l’élimination des substances interférantes. L’hypochlorite de sodium, le H2O2, le TCA et le PCA ont été évalués pour leur compatibilité avec le test U-2012 en utilisant le BSA comme protéine d’essai. Entre l’hypochlorite de sodium et l’H2O2, seul l’H2O2 était compatible car un précipité se formait en présence d’hypochlorite. Les protéines précipitées par le TCA ou le PCA peuvent être dosées par U-2012 après neutralisation adéquate de l’acide résiduel dans le culot. La supériorité du PCA sur le TCA pour la précipitation des protéines a été rapportée (24,25). En revanche, notre évaluation comparative a révélé des valeurs similaires de C50 pour le PCA (1,395) et le TCA (1,400). Nous avons préféré le PCA car il est facilement disponible sous forme de solution pré-fabriquée (70% v / v) et donc facilement dilué à la force requise. Le TCA est un solide hygroscopique difficile à peser précisément en raison de sa teneur en eau variable.
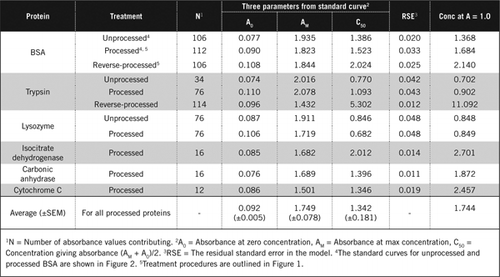
Il existe deux façons possibles de combiner PCA et H2O2. Pour les protéines « traitées », le traitement par PCA a été suivi d’un traitement par H2O2 et pour les protéines « traitées à l’envers », le traitement par H2O2 a précédé la précipitation par PCA. Les avantages de l’utilisation de protéines « transformées » étaient l’élimination d’un certain nombre de substances interférantes dans le surnageant et l’inactivation possible des enzymes protéolytiques lors de la préparation de l’échantillon. Ceci a été confirmé par le dosage de la trypsine et de la BSA traitées et inversées (voir tableau 1). Seuls les échantillons traités ont été utilisés pour déterminer la teneur réelle en protéines des échantillons biologiques colorés
Les traitements PCA et H2O2 d’échantillons colorés étaient nécessaires pour l’élimination des interférences dans le test U-2012. La précipitation acide seule des échantillons colorés n’a pas complètement éliminé les interférences. Avec tous les échantillons colorés, une certaine couleur a été jetée dans le surnageant, mais les pastilles ont également été colorées. La couleur a été éliminée des granulés par traitement H2O2. Des conditions alcalines étaient nécessaires à la fois pour une décoloration efficace par H2O2 (26) et pour le développement de la couleur par le réactif de Folin afin de s’assurer que les niveaux de protéines sont mesurés correctement. Bien que le NaOH et le KOH puissent tous deux fournir l’alcalinité requise, seul le NaOH était compatible avec le test U-2012. Un précipité s’est formé en présence de KOH. Dans les pastilles, le PCA a été neutralisé à l’aide de Na2CO3 et de NaOH(27). Du NaOH supplémentaire a été ajouté au cours de l’essai; le volume optimisé se situait entre 50 et 70 µL (60 µL étaient couramment utilisés); voir la figure 1.
La betterave, la myrtille et le vin rouge ont été décolorés avec 15 µL de H2O2 à 30% en prenant respectivement 0,5 et 2 h à 50 °C et à température ambiante. Vingt microlitres de H2O2 à 30% pendant 1 h à 50°C ont été utilisés pour faire face à des échantillons colorés plus forts. L’oxydation de substances telles que les sucres liés aux protéines par H2O2 à 50 ° C semble critique car le traitement à la température ambiante surestime la teneur en protéines. Dans le cas de la betterave, le traitement à 50 ° C a réduit l’estimation des protéines apparentes à 14% de l’estimation non traitée, alors que le traitement à la température ambiante n’a réduit que de moitié cette estimation.
Il ressort des dosages colorimétriques effectués après traitement au peroxyde d’hydrogène qu’une partie du H2O2 n’a pas été utilisée dans la décoloration. Dans un tel échantillon, la couleur finale du test de Lowry a été partiellement détruite. Il était donc nécessaire de détruire le peroxyde d’hydrogène restant avant le dosage des protéines. Il existe deux sources de H2O2 dans le test U-2012; H2O2 ajouté pour la décoloration et H2O2 présent comme contaminant dans le Triton X-100 (0,22%, Informations sur le produit: Triton X-100, www.sigmaaldrich.com ). Le peroxyde d’hydrogène est généralement dégradé par l’enzyme catalase. Cependant, le pH élevé du test protéique inactiverait les catalases connues. En outre, l’ajout de catalase entraînerait l’ajout de protéines supplémentaires. Nous avons choisi la destruction chimique du H2O2 à l’aide de pyruvate (28). La chimie de l’équation d’interaction pyruvate-H2O2 est bien établie (28,29). Le pyruvate détruit H2O2 à température ambiante selon la réaction suivante:

Le H2O2 résiduel dans la suspension du culot a été détruit par traitement avec du pyruvate de 0,9 M (concentration de 1,5x en H2O2) pendant 0,5 h à température ambiante. Pour contrer le H2O2 contaminant dans le Triton X-100, du pyruvate supplémentaire a également été ajouté dans le dosage des protéines (figure 1). L’ajout de pyruvate a donné une absorbance plus faible pour une ébauche non protéique. Nous suggérons que le contaminant peroxyde du Triton X-100 réagit avec l’acétonitrile de la solution-2, ce qui donne une capacité d’absorption légèrement supérieure.
L’interférence de couleur associée aux échantillons biologiques colorés ne peut pas simplement être prise en compte en effectuant un dosage protéique en l’absence du réactif de Foline. Les rapports calculés (Abs1-Abs2) / (Abs3-Abs4) indiquaient que l’interférence de la couleur de l’échantillon était la plus élevée pour le vin rouge (= 40) et moins pour la myrtille (= 6) et la betterave (= 2). Cette interférence s’est traduite par des estimations anormalement élevées des niveaux réels de protéines; par exemple, la concentration de protéines en utilisant des homogénats de betteraves non transformés et transformés (20,21 versus 2,89 mg de protéines / g de tissu, respectivement). En plus de l’interférence de couleur, le vin rouge et les homogénats de betterave et de myrtille sont susceptibles de contenir des substances qui réagiront avec le réactif de Folin dans l’essai U-2012 (e.g., petits peptides et sucres complexes). Ceux-ci ont été éliminés en précipitant sélectivement des protéines avec du PCA glacé à une concentration finale de 5% (Figure 1).
Courbes standard et leurs paramètres
Les courbes standard pour le BSA non traité et traité sont représentées à la figure 2. Les paramètres dérivés (A0, AM et C50) sont également énumérés dans le tableau 1 pour BSA et d’autres protéines.
Les résultats montrent que l’erreur type résiduelle dans le modèle est faible (0,012 à 0,048), ce qui indique que les données correspondent mieux à la tendance de l’hyperbole rectangulaire. Pour comparer les informations entre différentes protéines et leur traitement, les paramètres ont été convertis en concentration pour absorbance = 1,0 à 650 nm (colonne de droite du tableau 1).
Ces résultats montrent que la perte de protéines (par rapport aux protéines non transformées) dans les échantillons traités était inférieure à celle des échantillons traités à l’envers. Cette perte était plus apparente dans le cas de la trypsine et elle peut s’expliquer sur la base de son activité auto-catalytique lors du traitement inverse. Nous recommandons que le protocole « traité » (Matériel supplémentaire) ne soit suivi que pour les échantillons biologiques susceptibles de contenir des enzymes protéolytiques.
Dans le test de Lowry original (6) et sa version modifiée U-1988 (15), seule la portion linéaire de la courbe standard obtenue en traçant l’absorbance par rapport à la quantité de protéine a été utilisée pour la détermination quantitative de la protéine. Dans le test U-2012, nous utilisons les données plus efficacement en ajustant une équation d’hyperbole rectangulaire comme décrit dans la section Matériaux et méthodes conformément à Coakley et James (20).
Teneur en protéines de l’homogénat coloré
Les concentrations de protéines dans des échantillons inconnus ont été calculées par équation et par rapport à la norme BSA traitée et à la moyenne de toutes les protéines traitées énumérées dans le tableau 1. Ce dernier sera plus proche d’une estimation réelle pour des échantillons biologiques contenant un mélange de protéines. Nous avons estimé les quantités de protéines dans la myrtille et la betterave par rapport au vin rouge à environ 60 et 230 fois respectivement (tableau 2).
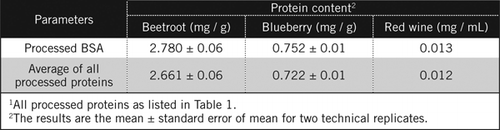
Comme le BSA, le vin rouge et 50% d’homogénats de betterave et de myrtille ont été traités par précipitation PCA et décoloration par H2O2 (figure 1). A ce stade, les échantillons biologiques ont été concentrés 40 fois pour le vin rouge et 4 fois pour la betterave et la myrtille. De même, le BSA (2 mg / mL) a également été concentré 4 fois à 8 mg/ mL. L’absorbance des échantillons colorés qui était proche de l’absorbance pour C50 (pour le BSA non traité) a été utilisée pour calculer la teneur en protéines, comme décrit par l’équation et.
En conclusion, l’essai U-2012 a utilisé des réactifs stables, a fourni une sensibilité améliorée (même pour les échantillons biologiques incolores) et a surmonté les interférences induites par la couleur pour les échantillons biologiques colorés. Le test U-2012 n’est pas limité à la partie linéaire de la réponse entre la concentration en protéines et l’absorbance et permet une utilisation plus efficace des données dans la région non linéaire grâce à un modèle de courbe hyperbolique rectangulaire adapté aux normes en utilisant des procédures simples dans Microsoft Excel.
Remerciements
Les auteurs remercient la Fondation pour la Recherche, la Science et la technologie de Nouvelle-Zélande pour son soutien financier (C06X0809).
Intérêts concurrents
Les auteurs ne déclarent aucun intérêt concurrent.
Données supplémentaires
Pour consulter les données supplémentaires qui accompagnent le présent article, veuillez visiter le site Web de la revue à l’adresse suivante : www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R.S. et P.L. Pedersen. 1985. Détermination des quantités de microgrammes de protéines en présence de milligrammes de lipides avec amido black 10B. Anal. Biochem. 150:97–104.Crossref, Medline, CAS, Google Scholar
- 2. Il s’agit de l’un des plus grands noms de la littérature française. 1949. Détermination des protéines sériques au moyen de la réaction de biuret. J. Biol. Chem. 177:751–766.Medline, CAS, Google Scholar
- 3. Smith, P.K., R.I. Krohn, G.T. Hermanson, A.K. Mallia, F.H. Gartner, M.D. Provenzano, E.K. Fujimoto, N.M. Goeke, et al.. 1985. Mesure de la protéine à l’aide d’acide bicinchoninique. Anal. Biochem. 150:76–85.Crossref, Medline, CAS, Google Scholar
- 4. Bradford, M.M. 1976. Une méthode rapide et sensible pour la quantification de quantités de microgrammes de protéines utilisant le principe de liaison protéine-colorant. Anal. Biochem. 72:248–254.Crossref, Medline, CAS, Google Scholar
- 5. Zor, T. et Z. Selinger. 1996. La linéarisation du test de la protéine de Bradford augmente sa sensibilité: études théoriques et expérimentales. Anal. Biochem. 236:302–308.Crossref, Medline, CAS, Google Scholar
- 6. Il s’agit de l’une des principales sources d’information sur la santé et la santé au travail. 1951. Mesure des protéines avec le réactif Folin phénol. J. Biol. Chem. 193:265–275.Medline, CAS, Google Scholar
- 7. 1979. Examen de la méthode de quantification de la protéine foline phénol de Lowry, Rosebrough, Farr et Randall. Anal. Biochem. 100:201–220.Crossref, Medline, CAS, Google Scholar
- 8. Sapan, C.V., R.L. Lundablad, et N.C. Prix. 1999. Techniques de dosage colorimétrique des protéines. Biotechnol. Appl. Biochem. 29:99–108.Medline, CAS, Google Scholar
- 9. Okutucu, B., A. Dınçer, Ö. Habib, et F. Zıhnıoglu. 2007. Comparaison de cinq méthodes de détermination de la concentration totale de protéines plasmatiques. J. Biochem. Biophys. Méthodes 70:709-711.Crossref, Medline, CAS, Google Scholar
- 10. Kresge, N., R.D. Simoni et R.L. Hill. 2005. L’article le plus cité dans l’histoire de l’édition: la détermination des protéines par Oliver H. Lowry. J. Biol. Chem. 25:280.Google Scholar
- 11. Il s’agit d’une espèce de poissons de la famille des Sauterelles, de la famille des Sauterelles et de la sous-famille des Sauterelles. 2010. Etude approfondie de la réactivité de diverses classes de composés vis-à-vis du réactif Folin-Ciocalteu. J. Agric. Chem alimentaire. 58:8139–8144.Crossref, Medline, CAS, Google Scholar
- 12. Eichberg, J. et L.C. Mokrasch. 1969. Interférence des lipides oxydés dans la détermination de la protéine par la procédure Lowry. Anal. Biochem. 30:386–390.Crossref, Medline, CAS, Google Scholar
- 13. Dulley, J.R. et P.A. pleurent. 1975. Une technique simple pour éliminer les interférences des détergents dans la méthode Lowry de détermination des protéines. Anal. Biochem. 64:136–141.Crossref, Medline, CAS, Google Scholar
- 14. Brillouet, J.-M., M.-P. Belleville et M. Moutounet. 1991. Complexes protéino-polysaccharidiques possibles dans les vins rouges. Être. J. Énol. Viti. 42:150–152.CAS, Google Scholar
- 15. Upreti, G.C., R.A. Ratcliff et P.C. Riches. 1988. Estimation des protéines dans les tissus contenant des niveaux élevés de lipides: modifications de la méthode Lowry de détermination des protéines. Anal. Biochem. 168:421–427.Crossref, Medline, CAS, Google Scholar
- 16. Upreti, G.C., C. Davis et J. Oliver. 1991. Préparation d’homogénéats représentatifs de tissus biologiques: effet du sel sur l’extraction des protéines. Anal. Biochem. 198:298–301.Crossref, Medline, CAS, Google Scholar
- 17. Smith, M.R., M.H. Penner, S.e. Bennett et A.T. Bakalinsky. 2011. Dosage Colorimétrique Quantitatif des Protéines Totales Appliqué au Vin Rouge Pinot Noir. J. Agric. Chem alimentaire. 59:6871–6876.Crossref, Medline, CAS, Google Scholar
- 18. Wigand, P., S. Tenzer, H. Schild et H. Decker. 2009. Analyse de la Composition Protéique du Vin Rouge en Comparaison avec les Vins Rosés et Blancs par Électrophorèse et Chromatographie Liquide à Haute Pression – Spectrométrie de Masse (HPLC-MS). J. Agric. Chem alimentaire. 57:4328–4333.Crossref, Medline, CAS, Google Scholar
- 19. Upreti, G.C., Y. Wang, A. Sharrock, N. Feisst, M. Davy et B. Jordan. 2009. Un test de protéines stables et sensibles (essai modifié U-2009) pour des échantillons biologiques colorés. ComBiol., Programme final de la Nouvelle-Zélande Décembre 2009. Université de Canterbury, Christchurch, Nouvelle-Zélande.Google Scholar
- 20. Il s’agit de l’un des plus grands ouvrages de l’histoire de l’art et de l’histoire de l’art. 1978. Une simple transformation linéaire pour la courbe d’étalonnage de la protéine Folin-Lowry à 1,0 mg/mL. Anal. Biochem. 85:90–97.Crossref, Medline, CAS, Google Scholar
- 21. Pinheiro, J.C. et D.M. Bates. 2000. Modèles à Effets mixtes dans les séries S et S-PLUS, Statistiques et Calcul. La société Springer – Verlag, New York, New York.Google Scholar
- 22. Équipe de Base de développement R. 2009. R : Un langage et un environnement pour le calcul statistique. R Fondation pour le Calcul statistique, Vienne, Autriche, ISBN 3-900051-07-0, URL http://www.R-project.org.Google Chercheur
- 23. Khachik, F., G.R. Beecher, J.T. Vanderslice et G. Sillon. 1988. Artefacts chromatographiques en phase liquide et distorsion maximale: Interaction échantillon-solvant dans la séparation des caroténoïdes. Anal. Chem. 60:807–811.Crossref, Medline, CAS, Google Scholar
- 24. Cernik, AA 1970. Détermination du plomb chélaté avec de l’acide éthylènediaminetétra-acétique dans le sang après précipitation de la protéine avec de l’acide perchlorique. Brit. J. Industrie Med. 27:40–42.Medline, CAS, Google Scholar
- 25. Moughan, P.J., A.J. Darragh, W.C. Smith et C.A. Butts. 1990. Les acides perchloriques et trichloroacétiques en tant que précipitants de protéines dans la digeste iléale endogène du rat. J. Sci. Agro-alimentaire. 52:13–21.Crossref, CAS, Google Scholar
- 26. Galbács, Z.M. et L.J. Csányi. 1983. Décomposition alcaline du peroxyde d’hydrogène. J. Chem. Soc., Dalton Trans. 11:2353–2357.Crossref, Google Scholar
- 27. Scopes, R.K. 1988. Purification des protéines: Principes et pratique, Deuxième Éd. La société Springer – Verlag New York Inc., New York, New York.Google Scholar
- 28. Upreti, G.C., K. Jensen, R. Munday, D.M. Duganzich, R. Vishwanath et J.F. Smith. 1998. Études sur l’activité oxydase des acides aminés aromatiques dans les spermatozoïdes ram: rôle du pyruvate en tant qu’antioxydant. Anim. Reprod. Sci. 51:275–287.Crossref, Medline, CAS, Google Scholar
- 29. Holleman, M.A.F. 1904. Avis sur l’action de l’eau oxygénée sur les acides α-cétoniques et sur les dicétones 1.2. Recl. Trav. Chim. Pays-Bas Belge 23:169–172.Crossref, CAS, Google Scholar