kvantitativa mått på fysiologiska egenskaper såsom enzymaktivitet uttrycks ofta som aktivitetsenheter per milligram protein. Även om många analyser har utvecklats för att mäta proteininnehåll, inklusive kolorimetriska analyser av Amido Black (1), Biuret (2), Bicinchonin Acid (3) och Coomassie Blue (4,5), Lowry-analysen (6) eller dess modifieringar (7,8) används oftare än andra analyser (9). Lowry-analysen är enkel, känslig och exakt och är den mest citerade (10) proceduren för kvantitativ proteinbestämning.
en mängd olika föreningar som reagerar med Folin-Ciocalteu fenol (Folins) reagens (11) är en källa till potentiell interferens i Lowry-och modifierade Lowry-proteinanalyser. Lyckligtvis är korrigeringar genom ett lämpligt ämne tillräckligt för de flesta föreningar (6,7) utom lipider (12), tvättmedel (13) och färgade ämnen (14). Svårigheter vid analys av proteiner i närvaro av lipider och tvättmedel (används vid solubilisering av fettvävnad, myelin och skelettmuskler) övervinnades av den modifierade Lowry-analysen (15; avses i detta dokument som u-1988-analysen, 16). Färginterferens vid bestämning av proteininnehållet i rött vin (14,17,18) övervanns genom att använda omfattande kromatografi. Ovanstående tillvägagångssätt är besvärligt och inte särskilt praktiskt för hantering av ett stort antal prover. Ingen av de kända proteinanalyserna var lämpliga för mätning av proteiner i färgade biologiska prover t. ex., färgade frukter och grönsaker, rött vin, pigmenterade mikrober och idisslare gall.
vår utveckling av u-2012-analysen från sina föregångare U-1988 och Lowry-analysen har uppnått tre stora fördelar (i) bekvämlighet genom stabilitet av reagensformuleringarna, (ii) mätning av protein i både färglösa och färgade biologiska prover utan att kompromissa med känsligheten och (iii) analys av proteiner vid mycket låga koncentrationer. Denna nya analys kommer att vara tillämplig på kvantitativ bestämning av protein i både färglösa och färgade biologiska provhomogenater, inklusive de som är rika på lipider (t.ex. avokado) och de som är svåra att homogenisera.
- material och metoder
- biologiska prover – rödbetor, blåbär och rött vin
- kemiska reagenser
- förbättringar av U-1988-analysen
- u-2012-analysen
- uppskattning av färgstörningar i u-2012-analysen
- standardkurvan och dess parametrar
- beräkning av proteininnehållet i homogenaten
- resultat och diskussion
- förbättringar i U-1988-analysen
- proteinuppskattning i färgade biologiska prover
- proteinutvinning
- eliminera störande ämnen
- standardkurvor och deras parametrar
- proteininnehåll i färgat homogenat
- bekräftelser
- konkurrerande intressen
- tilläggsdata
material och metoder
biologiska prover – rödbetor, blåbär och rött vin
rödbetor och blåbärshomogenater framställdes enligt beskrivningen i Tilläggsmaterialet. Rött vin krävde inte proteinutvinning före u-2012-analysen.
kemiska reagenser
alla kemiska reagenser, utom natriumhypoklorit och perklorsyra (PCA), erhölls från Sigma eller Sigma-Aldrich (St Louis, MO. USA). Natriumhypoklorit var från Acros Organic, New Jersey, USA. PCA erhölls från BDH (England).
förbättringar av U-1988-analysen
byte från karbonat till fosfatbuffert vid pH 12.0 förbättrade reagensstabiliteten och gav en liten ökning av känsligheten. Acetonitril infördes för att undvika tvättmedelsinducerade bubblor. NaOH ersatte KOH för att undvika utfällning i proteinanalysen. Dessutom förbättrades effektiviteten genom att kombinera olika komponenter i Lowry-reagenset i en reagensblandning.
u-2012-analysen
fullständig information om u-2012-analysen tillhandahålls i kompletterande Material. Protokollet, kort sammanfattat i Figur 1, beskriver bearbetningen av rött vin och homogenaten av rödbetor och blåbär, och inkluderar förbättringarna av U-1988-analysen. U-2012-analysen användes för obearbetade, bearbetade och omvända bearbetade (H2O2-behandling följt av TCA-eller PCA-utfällning) proteiner. Analyser utfördes på BSA, kolsyraanhydras, cytokrom C, isocitratdehydrogenas, lysozym och trypsin för utveckling av standardkurvor och i färgade biologiska prover. Bestämningen av proteiner i de biologiska proverna utfördes genom kalibrering till lämpliga standardkurvor.
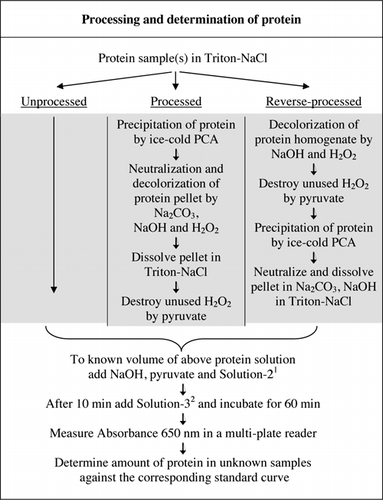
1solution-2 innehöll kopparsulfat (CuSO4.5h2o), Na-K-tartrat, SDS och acetonitril i 100 mM fosfatbuffert (pH 12,0). 2för lösning-3 späddes Folin-Ciocalteus fenolreagens 1:1 med avjoniserat vatten strax före användning.
uppskattning av färgstörningar i u-2012-analysen
Färginterferens bestämdes genom att jämföra absorptionsförmåga från bearbetade och obearbetade rödbetor, blåbär och rött vinprover både med och utan användning av Folins reagens som beskrivs i Figur 1. Förhållandet användes för att fastställa omfattningen av störningar, där Abs1 är absorption av obearbetade prover med Folins reagens; Abs2 är absorption av obearbetade prover utan Folins reagens; Abs3 är absorption av bearbetade prover med Folins reagens; och Abs4 är absorbans av bearbetade prover utan Folins reagens.
standardkurvan och dess parametrar
lösning – 1b och 1C som beskrivs i receptavsnittet i det bifogade Tilläggsmaterialet användes för utveckling av standardkurvorna. Koncentrationen av BSA och motsvarande absorbansvärden plottades med användning av en X-Y-spridningsgraf. Formen av denna graf (Figur 2) visar ett mättande svar vid högre koncentrationer med ett mycket begränsat initialt linjärt svar. Detta var en föredragen kurvform som rapporterats tidigare (20). Ursprungligen modellerades detta med hjälp av en exponentiell form (19) men senare studier visade att en rektangulär hyperbola gav en förbättrad anpassning till svaret, särskilt vid lägre koncentrationer. Denna senare form har nu standardiserats och följande treparameterekvation användes för att beskriva absorbansproteinkoncentrationsförhållandet:

Conc = proteinkoncentration,
A = absorbans vid Conc,
A0 = absorbans vid nollkoncentration,
AM= absorbans vid maximal koncentration,
C50= koncentration som ger absorbans
(AM + A0)/2.
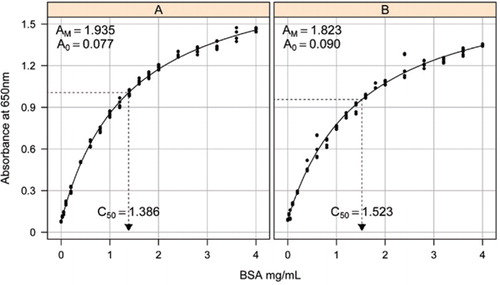
uppskattning av standardkurvan mellan absorbansen (650 nm) och koncentrationerna av BSA-protein gjordes på tre tekniska replikat till (A) den obearbetade proteinanalysen (parametrar A0= 0,077, Am=1,935, C50=1.386) och (B) den bearbetade proteinanalysen (parametrar A0=0,90, Am=1,823, C50=1,523). En rektangulär hyperbolamodell (ekvation ) monterades på de observerade data. Parameteruppskattningar identifieras i kurvorna med C50-uppskattningen på x-axeln (prickad svart linje).
Parameter A0 bestämdes experimentellt medan AM och C50 uppskattades med hjälp av Microsoft Excels Toolbox add-In Solver-funktion. En försöksuppsättning parametrar användes för att beräkna den modellerade absorbansen vid var och en av standardkoncentrationerna (Conc) med hjälp av ekvation . Solver beordrades sedan att minimera den återstående standardavvikelsen mellan den uppmätta och modellerade absorbansen för standarduppsättningen genom att justera AM och C50.
vi observerade förhållandet mellan absorbans och koncentration för att uppvisa en icke-linjär kurva över hela koncentrationsområdet, vilket sannolikt beror på en komponent av ljusspridning som ökar när koncentrationen av protein ökar vid mätning av absorbans. En dålig linjär passform vid låg absorbans rapporterades också av Coakley och James (20).
beräkning av proteininnehållet i homogenaten
analyser utfördes på bearbetat och obearbetat rött vin och homogenater av rödbetor och blåbär. BSA och andra proteinprover behandlades identiskt för lämpliga standardkurvor för att bestämma A0 -, AM-och C50-värden. Dessa parametrar användes sedan för att omvandla provabsorbans (A) till proteinkoncentration i varje homogenat med användning av:

eftersom ekvationen har en mättande form minskar känsligheten när absorbansen (A) och därmed koncentrationen ökar. Fel i proteinuppskattningar kan minimeras genom att justera koncentrationerna av homogenater i analysen så att de inte överstiger C50-värdet.
Homogenatkonc-värdet omvandlades sedan till vävnadsproteinkoncentration (Vävnadskonc i mg / g vävnad) med hjälp av följande formel:

där Homogenatkonc (i mg protein / mL) har korrigerats för eventuell förkoncentration eller spädning under analysen. Homogenatprocent var 100 g vävnad homogeniserad till en total volym av 200 mL (i vårt fall 50%).
i en separat studie monterades en rektangulär hyperbolamodell med användning av paketet non linear mixed effects (NLME) (21) i R (22) (Figur 2). Varje BSA-lösning, tillverkad oberoende i laboratoriet, modellerades som en slumpmässig effekt, med en gemensam A0 men olika AM-och C50-koefficienter. Detta modellerar hierarkin för biologiska provreplikater och tekniska analysreplikater.
resultat och diskussion
förbättringar i U-1988-analysen
begränsningen av U-1988 och Lowry-analysen är instabiliteten hos det karbonatbaserade reagenset. Karbonatbufferten (pH 11,4 vid 2% = 188,7 mM) i U-1988 ersattes med 40 mM fosfat vid pH-värden från 11,4 till 12,5. Initiala lutningar från standardkurvorna för proteinanalysen med användning av BSA vid 0,5 mg BSA/ mL och 1,0 mg BSA / mL beräknades. De initiala sluttningarna med fosfatbuffertar vid pH 11,4 och vid dess optimala pH 12,0 var 99 x10-6 och 197 x10-6respektivt. Lutningen för karbonatbufferten (pH 11,4) var 162 x10-6.Eftersom lutningsvärdet är en direkt indikation på analyskänslighet, fosfatbuffert (pH 12.0) valdes för att ersätta karbonatbuffert, vilket ger en 25% ökning av känsligheten.
större stabilitet uppnåddes genom ökad koncentration av fosfatbufferten till 100 mm. Den resulterande fosfat/CuSO4/Na-K-tartratlösningen var stabil vid rumstemperatur i två veckor, betydligt längre än karbonat/CuSO4/Na-K-tartratlösningen, som måste beredas dagligen före proteinanalys. För alla framtida experiment användes 100 mM fosfat (pH 12.0) för att förbereda CuSO4/Na-K-tartratlösningen. Vi tror att ersättning av karbonat med fosfat kommer att förbättra bekvämligheten med U-2012-analysen.
Tvättmedelsinducerade bubblor blir en viktig felkälla vid absorptionsmätningar vid användning av en flerbrunnsplattaläsare (inte ett problem med kyvetter). Dessa bubblor reducerades avsevärt genom tillsats av ett antal polära lösningsmedel (t.ex. aceton, acetonitril, etanol och metanol). Acetonitril, den mest polära av dessa lösningsmedel (23) valdes för dess effektivitet och ingår i lösning-2 (Se figur 1 bildtext och recept avsnitt av kompletterande Material).
fosfatbuffert, CuSO4, Na-K-tartrat, SDS och acetonitril kan tillsättas individuellt och ordningen för deras tillsats påverkar inte den resulterande absorbansen. Att använda en förblandad lösning förbättrar emellertid bekvämligheten ytterligare, särskilt när ett stort antal prover ska analyseras. Vi grupperade därför dessa komponenter i analysblandningen i lösning – 2 (Figur 1). En sådan förblandad lösning var inte möjlig för den ursprungliga Lowry-analysen (6) på grund av karbonatlösningens instabilitet. Försöket att inkludera lösning – 3 i lösning-2 resulterade i dramatisk minskning av utvecklingen av blå färg och ansågs inte vidare.
proteinuppskattning i färgade biologiska prover
proteinutvinning
proteiner från rödbetor och blåbär extraherades i Triton X-100-NaCl-lösning med mild homogenisering. Sådana homogenater behåller sina enzymaktiviteter (15). Denna extraktion krävdes inte för rött vin.
eliminera störande ämnen
för färgade prover är det nödvändigt att avlägsna störningen på grund av den inneboende provfärgen och andra icke-proteinämnen som reagerar med proteinreagensen före kolorimetrisk proteinanalys. Nyheten med U-2012 är att utforma ett avfärgningsprotokoll kompatibelt med en kolorimetrisk proteinanalys.
avfärgning av färgade pigment med natriumhypoklorit eller H2O2 och selektiv utfällning av proteiner med PCA eller TCA övervägdes för avlägsnande av störande ämnen. Natriumhypoklorit, H2O2, TCA och PCA utvärderades för deras kompatibilitet med U-2012-analysen med användning av BSA som testprotein. Mellan natriumhypoklorit och H2O2 var endast H2O2 kompatibel eftersom en fällning bildades i närvaro av hypoklorit. Proteiner utfällda av TCA eller PCA kan analyseras av U-2012 efter adekvat neutralisering av restsyra i pelleten. Överlägsenheten av PCA över TCA för proteinutfällning har rapporterats (24,25). Däremot avslöjade i vår jämförande utvärdering liknande C50-värden för PCA (1.395) och TCA (1.400). Vi föredrog PCA eftersom den är lätt tillgänglig som en färdig lösning (70% v/v) och därför lätt utspädd till önskad styrka. TCA är ett hygroskopiskt fast ämne som är svårt att väga exakt på grund av dess varierande vatteninnehåll.
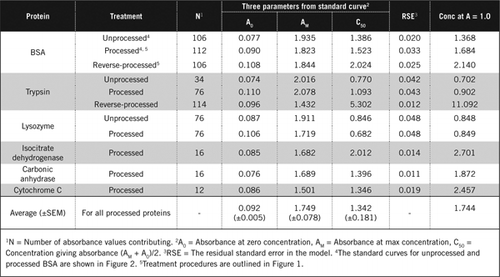
det finns två möjliga sätt att kombinera PCA och H2O2. För” bearbetade ”proteiner följdes PCA-behandling av H2O2-behandling och för” omvänd bearbetat ” protein föregick H2O2-behandling PCA-utfällning. Fördelarna med att använda’ bearbetat ’ protein var avlägsnandet av ett antal störande ämnen i supernatanten och möjlig inaktivering av proteolytiska enzymer under provberedningen. Detta bekräftades genom analys av bearbetat och omvänd bearbetat trypsin och BSA (se Tabell 1). Endast bearbetade prover användes för att bestämma det faktiska proteininnehållet i färgade biologiska prover
både PCA-och H2O2-behandlingar av färgade prover var nödvändiga för eliminering av störningar i u-2012-analys. Enbart syrautfällning av de färgade proverna avlägsnade inte störningen helt. Med alla färgade prover kastades viss färg i supernatanten, men pelletsna färgades också. Färg eliminerades från pelletsna genom H2O2-behandling. Alkaliska förhållanden krävdes för både effektiv avfärgning av H2O2 (26) och färgutvecklingen av Folins reagens för att säkerställa att proteinnivåerna mäts korrekt. Även om både NaOH och KOH kunde ge den erforderliga alkaliniteten, var endast NaOH kompatibel med U-2012-analysen. En fällning bildades i närvaro av KOH. I pellets neutraliserades PCA med användning av Na2CO3 och NaOH (27). Ytterligare NaOH tillsattes under analysen; den optimerade volymen var mellan 50 och 70 kg (60 kg användes rutinmässigt); se Figur 1.
rödbeta, blåbär och rött vin avfärgades med 15 occyl på 30% H2O2 som tog 0,5 och 2 h vid 50 oc C respektive rumstemperatur. Tjugo mikroliter av 30% H2O2 för 1 h vid 50 kcal C användes för att klara av starkare färgade prover. Oxidation av ämnen som sockerarter som är bundna till proteiner med H2O2 vid 50 kcal C verkar kritisk eftersom rumstemperaturbehandling överskattar proteininnehållet. När det gäller rödbetor minskade bearbetningen med 50 CCG den skenbara proteinuppskattningen till 14% av de obearbetade, medan bearbetningen vid rumstemperatur endast halverade den uppskattningen.
det var uppenbart från de kolorimetriska analyserna som utfördes efter väteperoxidbehandling att vissa H2O2 inte användes vid avfärgning. I ett sådant prov förstördes slutfärgen på Lowry-analysen delvis. Det var därför nödvändigt att förstöra den återstående väteperoxiden före proteinanalys. Det finns två källor till H2O2 i u-2012-analysen; H2O2 tillsatt för avfärgning och H2O2 närvarande som en förorening i Triton X-100 (0.22%, produktinformation: Triton X-100, www.sigmaaldrich.com). väteperoxid bryts vanligen ned av enzymet katalas. Emellertid skulle det höga pH-värdet av proteinanalysen inaktivera kända katalaser. Dessutom skulle tillsats av katalas leda till tillsats av extra protein. Vi valde kemisk förstörelse av H2O2 med pyruvat (28). Kemin för pyruvat-H2O2-interaktionsekvationen är väl etablerad (28,29). Pyruvat förstör H2O2 vid rumstemperatur enligt följande reaktion:

återstående H2O2 i pelletsuspensionen förstördes genom behandling med 0,9 m pyruvat (1,5 x koncentration av H2O2) under 0,5 h vid rumstemperatur. För att motverka den förorenande H2O2 i Triton X-100 tillsattes också extra pyruvat i proteinanalysen (Figur 1). Tillsatsen av pyruvat gav lägre absorbans för ett icke-proteinämne . Vi föreslår att peroxidföroreningen i Triton X-100 reagerar med acetonitril i lösning-2, vilket ger något högre absorptionsförmåga.
färginterferensen associerad med de färgade biologiska proverna kan inte bara beaktas genom att köra en proteinanalys i frånvaro av Folins reagens. De beräknade förhållandena (Abs1-Abs2) / (Abs3-Abs4) indikerade att störningar från provfärg var den högsta för rött vin (=40) och mindre för blåbär (=6) och rödbetor (=2). Denna störning översattes till de onormalt höga uppskattningarna av sanna proteinnivåer; till exempel koncentrationen av protein med användning av obearbetade och bearbetade rödbetorhomogenater (20, 21 mot 2, 89 mg protein / g vävnad). Förutom färginterferens kommer rött vin och homogenater av rödbetor och blåbär sannolikt att innehålla ämnen som kommer att reagera med Folins reagens i u-2012-analysen (e.g., små peptider och komplexa sockerarter). Dessa avlägsnades genom selektivt utfällning av proteiner med iskall PCA vid en slutlig koncentration av 5% (Figur 1).
standardkurvor och deras parametrar
Standardkurvor för obearbetad och bearbetad BSA visas i Figur 2. De härledda parametrarna (A0, AM och C50) listas också i Tabell 1 för BSA och andra proteiner.
resultaten visar att det återstående standardfelet i modellen är lågt (0,012 till 0,048) vilket indikerar bättre passform av data till den rektangulära hyperboltrenden. För att jämföra information mellan olika proteiner och deras bearbetning omvandlades parametrarna till koncentrationen för absorbans = 1,0 vid 650 nm (höger kolumn i Tabell 1).
dessa resultat visar att förlust av protein (jämfört med obearbetat protein) i bearbetade prover var mindre än de omvända bearbetade proverna. Denna förlust var tydligare i fallet med trypsin och det kan förklaras på grundval av dess auto-katalytiska aktivitet under omvänd bearbetning. Vi rekommenderar att det bearbetade protokollet (tilläggsmaterial) endast följs för biologiska prover som sannolikt innehåller proteolytiska enzymer.
i den ursprungliga Lowry-analysen (6) och dess modifierade version U-1988 (15) användes endast den linjära delen av standardkurvan erhållen genom att plotta absorbansen mot mängden protein vid kvantitativ bestämning av protein. I u-2012-analysen använder vi data mer effektivt genom att montera en rektangulär hyperbolaekvation som beskrivs i avsnittet material och metoder i linje med Coakley och James (20).
proteininnehåll i färgat homogenat
proteinkoncentrationer i okända prover beräknades genom ekvation och mot bearbetad BSA-standard och genomsnittet av alla bearbetade proteiner listade i Tabell 1. Den senare kommer att vara närmare en sann uppskattning för biologiska prover som innehåller en blandning av proteiner. Vi uppskattade mängden protein i blåbär och rödbetor i förhållande till rött vin som ungefär 60 respektive 230 gånger (tabell 2).
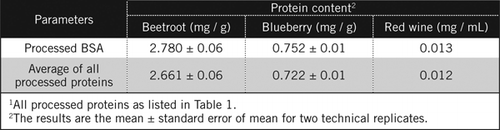
liksom BSA bearbetades rött vin och 50% homogenater av rödbetor och blåbär genom PCA-utfällning och avfärgning med H2O2 (Figur 1). I detta skede koncentrerades de biologiska proverna 40 gånger för rött vin och 4 gånger för rödbetor och blåbär. På samma sätt koncentrerades BSA (2 mg/mL) också 4 gånger till 8 mg/mL. Absorbansen hos de färgade proverna som var nära absorbansen för C50 (för obearbetad BSA) användes för att beräkna proteininnehållet, såsom beskrivits av ekvation och .
Sammanfattningsvis har u-2012-analysen använt stabila reagens, tillhandahållit förbättrad känslighet (även för färglösa biologiska prover) och övervunnit färginducerad interferens för färgade biologiska prover. U-2012-analysen är inte begränsad till den linjära delen av svaret mellan proteinkoncentration och absorbans och gör effektivare användning av data i den icke-linjära regionen genom en rektangulär hyperbolisk kurvmodell anpassad till standarderna med enkla procedurer inom Microsoft Excel.
bekräftelser
författare erkänner Stiftelsen för forskning, vetenskap och teknik Nya Zeeland för ekonomiskt stöd (C06X0809).
konkurrerande intressen
författarna förklarar inga konkurrerande intressen.
tilläggsdata
för att se tilläggsdata som följer med detta papper, besök tidskriftens webbplats på: www.future-science.com/doi/suppl/10.2144/000113818
- 1. Kaplan, R. S. och P. L. Pedersen. 1985. Bestämning av mikrogrammängder protein i närvaro av milligramnivåer av lipid med amido black 10B. Anal. Biochem. 150:97–104.Crossref, Medline, CAS, Google Scholar
- 2. Gornall, A. G., C. J. Bardawill och M. M. David. 1949. Bestämning av serumproteiner med hjälp av biuret-reaktionen. J. Biol. Chem. 177:751–766.Medline, CAS, Google Scholar
- 3. Han är en av de mest kända och mest kända i världen.. 1985. Mätning av protein med användning av bicinkoninsyra. Anal. Biochem. 150:76–85.Crossref, Medline, CAS, Google Scholar
- 4. Bradford, M. M. 1976. En snabb och känslig metod för kvantifiering av mikrogrammängder protein med principen om proteinfärgbindning. Anal. Biochem. 72:248–254.Crossref, Medline, CAS, Google Scholar
- 5. Zor, T. och Z. Selinger. 1996. Linjärisering av Bradford – proteinanalysen ökar dess känslighet: teoretiska och experimentella studier. Anal. Biochem. 236:302–308.Crossref, Medline, CAS, Google Scholar
- 6. Lowry, O. H., N. J. Rosbrough, A. L. Farr och R. J. Randall. 1951. Proteinmätning med Folin-fenolreagenset. J. Biol. Chem. 193:265–275.Medline, CAS, Google Scholar
- 7. Peterson, G. L. 1979. Granskning av kvantifieringsmetoden för folin fenolprotein för Lowry, Rosebrough, Farr och Randall. Anal. Biochem. 100:201–220.Crossref, Medline, CAS, Google Scholar
- 8. Sapan, C. V., R. L. Lundablad och N. C. Price. 1999. Kolorimetriska proteinanalystekniker. Bioteknol. Appl. Biochem. 29:99–108.Medline, CAS, Google Scholar
- 9. Okutucu, B., A. D. Habib,och F. Z. 2007. Jämförelse av fem metoder för bestämning av total plasmaproteinkoncentration. J. Biochem. Biophys. Metoder 70: 709-711.Crossref, Medline, CAS, Google Scholar
- 10. Kresge, N., R. D. Simoni och R. L. Hill. 2005. Det mest citerade papperet i publiceringshistoria: proteinbestämning av Oliver H. Lowry. J. Biol. Chem. 25:280.Google Scholar
- 11. Everette, J. D., Q. M. Bryant, A. M. Green, Y. A. Abbey, G. W. Wangila och R. B. Walker. 2010. Grundlig studie av reaktivitet hos olika föreningsklasser mot Folin-Ciocalteu-reagenset. J. Agric. Mat Chem. 58:8139–8144.Crossref, Medline, CAS, Google Scholar
- 12. Eichberg, J. och L. C. Mokrasch. 1969. Interferens av oxiderade lipider vid bestämning av protein genom Lowry-förfarandet. Anal. Biochem. 30:386–390.Crossref, Medline, CAS, Google Scholar
- 13. Dulley, J. R. och P. A. Grieve. 1975. En enkel teknik för att eliminera störningar av tvättmedel i Lowry-metoden för proteinbestämning. Anal. Biochem. 64:136–141.Crossref, Medline, CAS, Google Scholar
- 14. Brillouet, J.-M., M.-P. Belleville och M. Moutounet. 1991. Möjliga protein-polysackaridkomplex i röda viner. Är. J. Enol. Vitic. 42:150–152.CAS, Google Scholar
- 15. Upreti, G. C., R. A. Ratcliff och P. C. Riches. 1988. Proteinuppskattning i vävnader som innehåller höga nivåer av lipid: modifieringar av Lowry-metoden för proteinbestämning. Anal. Biochem. 168:421–427.Crossref, Medline, CAS, Google Scholar
- 16. Upreti, G. C., C. Davis och J. Oliver. 1991. Framställning av representativa homogenater av biologiska vävnader: effekt av salt på proteinutvinning. Anal. Biochem. 198:298–301.Crossref, Medline, CAS, Google Scholar
- 17. Smith, M. R., M. H. Penner, S. E. Bennett och A. T. Bakalinsky. 2011. Kvantitativ kolorimetrisk analys för totalt Protein applicerat på rött vin Pinot Noir. J. Agric. Mat Chem. 59:6871–6876.Crossref, Medline, CAS, Google Scholar
- 18. Wigand, P., S. Tenzer, H. Schild och H. Decker. 2009. Analys av proteinsammansättning av rött vin i jämförelse med ros-och vita viner med hjälp av elektrofores och högtrycksvätskekromatografi-masspektrometri (HPLC-MS). J. Agric. Mat Chem. 57:4328–4333.Crossref, Medline, CAS, Google Scholar
- 19. Upreti, G. C., Y. Wang, A. Sharrock, N. Feisst, M. Davy och B. Jordanien. 2009. En stabil och känslig proteinanalys (u-2009 modifierad analys) för färgade biologiska prover. ComBiol., Nya Zeelands Slutprogram December 2009. University of Canterbury, Christchurch, Nya Zeeland.Google Scholar
- 20. Coakley, W. T. och C. J. James. 1978. En enkel linjär transformation för Folin-Lowry – proteinkalibreringskurvan till 1,0 mg/mL. Anal. Biochem. 85:90–97.Crossref, Medline, CAS, Google Scholar
- 21. Pinheiro, J. C. och D. M. Bates. 2000. Blandade effekter modeller i S och S-PLUS, statistik och Datorserier. Springer-Verlag, New York, NY.Google Scholar
- 22. R Utveckling Core Team. 2009. R: ett språk och en miljö för statistisk databehandling. R Foundation for Statistical Computing, Wien, Österrike, ISBN 3-900051-07-0, URL http://www.R-project.org.Google Scholar
- 23. Khachik, F., G. R. Beecher, J. T. Vanderslice och G. Fåra. 1988. Vätskekromatografiska artefakter och toppförvrängning: interaktion mellan prov och lösningsmedel vid separation av karotenoider. Anal. Chem. 60:807–811.Crossref, Medline, CAS, Google Scholar
- 24. Cernik, A. A. 1970. Bestämning av blykelaterad med etylendiamintetra-ättiksyra i blod efter utfällning av protein med perklorsyra. Britt. J. Industri Med. 27:40–42.Medline, CAS, Google Scholar
- 25. Moughan, P. J., A. J. Darragh, W. C. Smith och C. A. Butts. 1990. Perklorsyra och triklorättiksyror som utfällningar av protein i endogen ileal digesta från råtta. J. Sci. Mat Agric. 52:13–21.Crossref, CAS, Google Scholar
- 26. Galb, Z. M. och L. J. CS. 1983. Alkaliinducerad sönderdelning av väteperoxid. J. Chem. Soc. Dalton Trans. 11:2353–2357.Crossref, Google Scholar
- 27. Scopes, R. K. 1988. Proteinrening: principer och övning, andra Ed. Springer-Verlag New York Inc., New York, NY.Google Scholar
- 28. Upreti, G. C., K. Jensen, R. Munday, D. M. duganzich, R. Vishwanath och J. F. Smith. 1998. Studier av aromatisk aminosyraoxidasaktivitet i RAM-spermatozoa: pyruvats roll som en antioxidant. Anim. Reprod. Sci. 51:275–287.Crossref, Medline, CAS, Google Scholar
- 29. Holleman, M. A. F. 1904. Lägg märke till sur l ’action de l’ Eau oxygenee sur les syror sackaros-cetoniques et sur les dicetones 1.2. Recl. Trav. Chim. Pays-Bas Belg. 23:169–172.Crossref, CAS, Google Scholar